
Platelets and Hemostasis
By the conclusion of this exhaustive guide, you will be deeply conversant with:
- The delicate physiological balance of Hemostasis (preventing hemorrhage vs. preventing thrombosis).
- The morphology, lifecycle, and critical functions of Platelets in Primary Hemostasis.
- The enzymatic cascade of Secondary Hemostasis, mastering both the Traditional and Cell-Based models.
- The body's natural Anticoagulant and Fibrinolytic systems.
- The interpretation of Hemostasis Laboratory Tests (PT, aPTT, INR, D-Dimer).
- The pathophysiology and clinical presentation of Bleeding and Thrombotic Disorders.
I. Introduction to Hemostasis
Hemostasis is the highly regulated, dynamic physiological process that halts bleeding at the site of a vascular injury while simultaneously maintaining normal, fluid blood flow elsewhere in the circulatory system. It represents an exquisite biological balancing act.
If the hemostatic balance tips in one direction, hemorrhage (excessive, life-threatening bleeding) occurs. If it tips in the opposite direction, thrombosis (inappropriate, dangerous blood clotting inside intact vessels) occurs, leading to conditions like myocardial infarctions (heart attacks) and ischemic strokes.
The process involves continuous interactions between three primary components:
- The Blood Vessel Wall (Endothelium): Healthy endothelium prevents clotting; injured endothelium strongly initiates it.
- Platelets (Thrombocytes): Cellular fragments that create the initial physical plug.
- The Coagulation Cascade: Plasma proteins that form a biological "cement" to solidify the plug.
II. Platelets
Platelets (thrombocytes) are small, anucleated (lacking a nucleus) cell fragments that play the central role in Primary Hemostasis—the rapid formation of an initial, temporary platelet plug at the site of vascular injury.
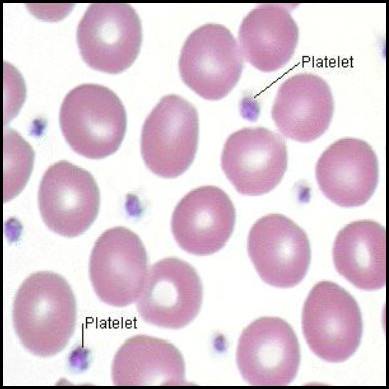
1. Morphology and Physical Traits
- Size and Shape: They are tiny, measuring only 2-4 µm in diameter. When resting/inactive, they are discoid (lens-shaped) to flow smoothly through capillaries. Upon activation, they undergo a massive conformational change, becoming spherical and shooting out long, spiky pseudopods (finger-like projections) to enhance their surface area and stickiness.
- Anucleated Nature: Because they lack a nucleus, platelets cannot transcribe DNA or synthesize new proteins. This is clinically vital: if a drug (like Aspirin) permanently disables a platelet's enzyme, that platelet is disabled for the rest of its life because it cannot build replacement enzymes.
- Lifespan: Highly limited, living only 7 to 10 days in circulation before being destroyed by macrophages in the spleen and liver.
2. The Platelet Membrane Receptors
The platelet membrane is studded with critical glycoprotein (GP) receptors that act as biological velcro, allowing the platelet to stick to injured tissue and to other platelets.
- GP Ib/IX/V Complex: Binds to von Willebrand Factor (vWF), which coats the exposed collagen of a damaged blood vessel.
- GP Ia/IIa Complex: Binds directly to exposed subendothelial Collagen.
- GP IIb/IIIa Complex: The most abundant receptor. When activated, it binds to Fibrinogen, which acts as a bridge to connect multiple platelets together. (Clinical Example: Drugs like Abciximab or Tirofiban are GP IIb/IIIa inhibitors used during heart procedures to prevent platelets from linking together).
The Cytoplasmic Payload
The platelet cytoplasm contains highly specialized granules that release chemicals to amplify the clotting response.
- Alpha-granules (The Proteins): Contain large proteins essential for adhesion and healing. Includes Fibrinogen, von Willebrand factor (vWF), Platelet factor 4 (PF4), Platelet-Derived Growth Factor (PDGF), and P-selectin.
- Dense/Delta-granules (The Activators): Contain smaller, non-protein molecules. Mnemonic: SAC. Serotonin (causes vasoconstriction), ADP/ATP (powerful platelet activators), and Calcium (essential for the coagulation cascade).
- Lysosomes: Contain hydrolytic enzymes for digesting extracellular material.
3. Formation of Platelets (Thrombopoiesis)
Platelet production occurs entirely in the bone marrow and is heavily regulated by a hormone called Thrombopoietin (TPO), which is primarily synthesized in the liver.
- Origin: Hematopoietic Stem Cells (HSCs) differentiate into the Common Myeloid Progenitor (CMP).
- Megakaryoblast Stage: The progenitor cell undergoes endoreduplication (DNA replicates repeatedly, but the cell never divides). The cell becomes massively polyploid (containing up to 64 copies of DNA).
- Megakaryocyte Stage: The resulting cell is the largest in the bone marrow (up to 100 µm) with a bizarre, highly lobulated nucleus.
- Platelet Release: The megakaryocyte extends long, ribbon-like "proplatelets" directly into the bone marrow sinusoidal capillaries. The sheer physical force of flowing blood fragments these extensions into thousands of individual platelets (about 1,000-3,000 per megakaryocyte).
TPO Regulation (Negative Feedback): TPO constantly stimulates megakaryocytes. Platelets floating in the blood have receptors that bind to and destroy TPO. Therefore, if platelet counts are high, all TPO is absorbed, and production slows down. If platelet counts drop (e.g., severe bleeding), free TPO levels rise, stimulating the marrow to produce more.
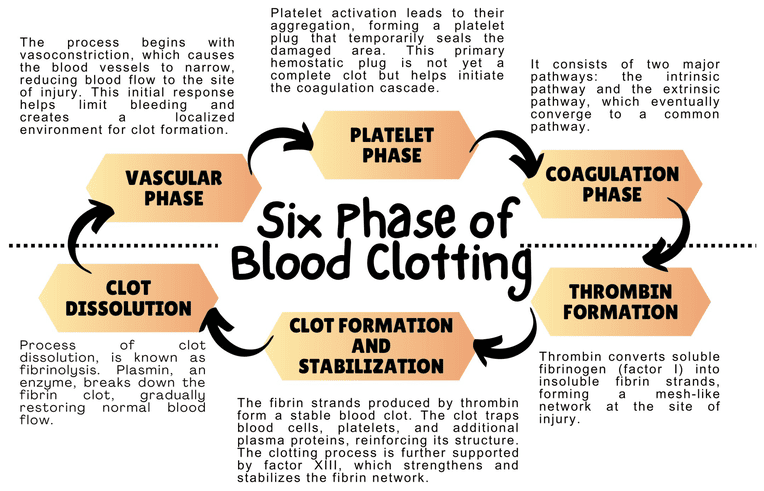
III. The Steps of Primary Hemostasis
When a blood vessel is damaged, exposing the underlying collagen and connective tissue, platelets execute a rapid, four-step response to plug the hole.
Step 1: Adhesion
- Vascular injury exposes subendothelial collagen.
- Endothelial cells release von Willebrand factor (vWF), which unfurls and sticks to the collagen.
- Platelets utilize their GP Ib receptors to bind to the vWF. Think of vWF as a highly adhesive glue connecting the damaged wall to the platelet. Direct binding to collagen also occurs via GP Ia/IIa.
Step 2: Activation
- Once tethered to the wall, platelets undergo a massive shape change (from smooth discs to spiky spheres) and "degranulate" (dump their alpha and dense granules into the blood).
- Key Molecules Released/Synthesized:
- ADP: A potent activator that calls thousands of other platelets to the area.
- Thromboxane A2 (TxA2): Synthesized rapidly inside the platelet via the COX-1 enzyme pathway. TxA2 is a violent vasoconstrictor and powerful platelet aggregator.
- Serotonin: Causes further vascular spasm to reduce blood loss.
Clinical Pharmacology: Aspirin Mechanism
Aspirin works as a blood thinner by irreversibly inhibiting the Cyclooxygenase-1 (COX-1) enzyme inside platelets. Without COX-1, the platelet cannot synthesize Thromboxane A2 (TxA2). Without TxA2, platelet activation and aggregation are severely crippled. Because platelets lack a nucleus, they cannot build new COX-1; therefore, the platelet is permanently disabled for its entire 7-10 day lifespan. This is why low-dose aspirin is given to prevent heart attacks.
Step 3: Aggregation
- Activation causes the platelet's GP IIb/IIIa receptors to change shape and become highly receptive.
- Plasma Fibrinogen binds to the GP IIb/IIIa receptors on adjacent platelets, acting as a molecular bridge.
- Thousands of platelets link together, forming the initial Primary Hemostatic Plug.
Step 4: Procoagulant Activity
- Activated platelets flip their cell membranes inside out, exposing a negatively charged lipid called phosphatidylserine on their outer surface.
- This negatively charged surface acts as the physical "workbench" where the enzymes of the Coagulation Cascade will assemble and function.
IV. Secondary Hemostasis (The Coagulation Cascade)
While the primary platelet plug is an excellent immediate seal, it is weak and friable. It cannot withstand the high-pressure pumping of arterial blood. Secondary Hemostasis involves a series of enzymatic reactions in the plasma that ultimately generate Fibrin—a tough, insoluble protein mesh that wraps around and solidifies the platelet plug.
We analyze this cascade using two models: the Traditional Model (used for understanding lab tests) and the Cell-Based Model (how it actually happens in the human body).
A. The Traditional Model (The Y-Shaped Pathway)
This model divides coagulation into the Extrinsic, Intrinsic, and Common pathways.
The "Initiator"
Activated rapidly by external trauma to the blood vessel.
- Step 1: Injury exposes Tissue Factor (TF), a protein normally hidden on smooth muscle cells and fibroblasts beneath the endothelium.
- Step 2: Circulating Factor VII binds to TF to form the TF-VIIa complex.
- Step 3: This complex directly activates Factor X (to Xa) and Factor IX (to IXa).
The "Amplifier"
Activated when blood comes into contact with negatively charged surfaces (like exposed collagen or glass in a test tube).
- Step 1: Factor XII is activated to XIIa.
- Step 2: XIIa activates Factor XI to XIa.
- Step 3: XIa activates Factor IX to IXa.
- Step 4 (The Tenase Complex): Factor IXa combines with cofactor VIIIa (and Calcium) on the platelet surface. This complex aggressively activates Factor X to Xa.
3. The Common Pathway
Both the Extrinsic and Intrinsic pathways converge at the activation of Factor X.
- Step 1 (The Prothrombinase Complex): Activated Factor X (Xa) combines with cofactor Va (and Calcium) on the platelet phospholipid surface.
- Step 2 (The Central Event): The Prothrombinase Complex takes Prothrombin (Factor II) and cleaves it into the highly active, powerful enzyme Thrombin (Factor IIa).
- Step 3 (The Role of Thrombin): Thrombin is the master regulator. It does multiple things simultaneously:
- Converts soluble Fibrinogen (Factor I) into insoluble Fibrin monomers.
- Activates Factor XIII (the cross-linker).
- Feeds back to massively activate Factors V, VIII, and XI (creating a violent positive feedback loop).
- Strongly activates more platelets.
- Step 4 (Stabilization): Fibrin monomers link together into a weak polymer. Finally, Factor XIIIa (a transglutaminase enzyme) acts like biological sewing thread, covalently cross-linking the fibrin strands into a highly stable, indestructible mesh.
B. The Cell-Based Model of Coagulation
In living humans, coagulation doesn't happen in isolated pathways; it happens directly on the surfaces of cells in three overlapping phases:
- Initiation Phase: Occurs on Tissue Factor-bearing cells outside the vessel. TF binds VIIa, activating small amounts of X and IX. This generates a tiny "Thrombin Spurt," which is not enough to clot blood, but enough to sound the alarm.
- Amplification Phase: The "Thrombin Spurt" activates local platelets and circulating cofactors (V, VIII, XI), priming the environment for a massive reaction.
- Propagation Phase: Occurs exclusively on the surface of activated platelets. The Tenase complex (IXa/VIIIa) and Prothrombinase complex (Xa/Va) assemble on the platelets, generating a massive "Thrombin Burst." This burst rapidly converts fibrinogen to a robust fibrin clot.
Vitamin K and Calcium
- Vitamin K-Dependent Factors: Factors II, VII, IX, X, Protein C, and Protein S all require Vitamin K for synthesis in the liver. Vitamin K adds a carboxyl group to these proteins, allowing them to bind to Calcium.
Pharmacology Note: Warfarin (Coumadin) is an oral blood thinner that poisons the Vitamin K recycling enzyme in the liver, shutting down the production of these factors. - Calcium (Factor IV): Calcium acts as the bridge connecting the coagulation factors to the negatively charged surface of the activated platelet. Without Calcium, blood cannot clot. (This is why blood donation bags contain EDTA or Citrate—chemicals that bind all the Calcium, keeping the blood liquid in the bag).
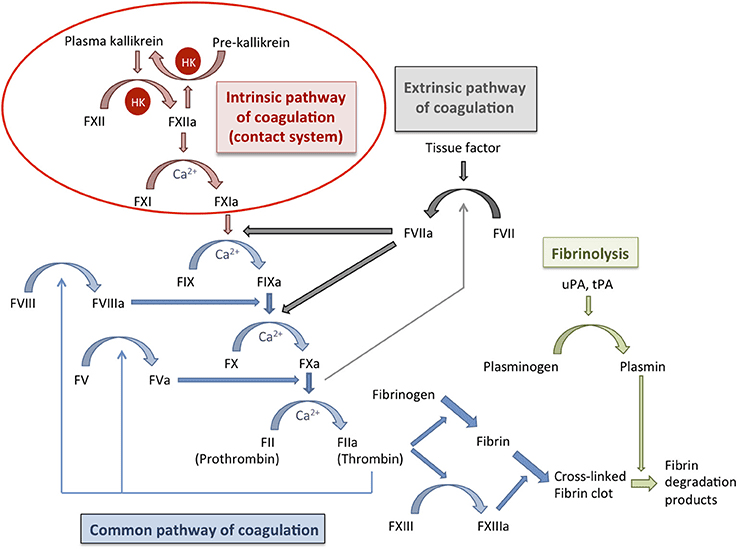
V. Regulation of Clotting and Fibrinolysis
Hemostasis requires aggressive regulation. If the coagulation cascade was left unchecked, a tiny cut on your finger would cause your entire circulatory system to clot solid. The body uses anticoagulation systems to restrict the clot strictly to the site of injury, and Fibrinolysis to dissolve the clot once healing is complete.
A. Natural Anticoagulation Systems
Mechanism: A major plasma protein that chemically binds to and destroys several activated factors, primarily Thrombin (IIa) and Factor Xa.
Pharmacological Enhancement: By itself, Antithrombin acts slowly. However, when it binds to Heparin (a drug) or heparan sulfate (found naturally on healthy blood vessels), its action is accelerated 1,000-fold. This is exactly how Heparin acts as a rapid IV blood thinner in hospitals.
Mechanism: When healthy, uninjured endothelium encounters stray Thrombin, a receptor called Thrombomodulin captures the Thrombin. This complex activates Protein C into Activated Protein C (APC).
Action: APC, assisted by Protein S, acts as a biological assassin, seeking out and destroying the essential cofactors Va and VIIIa. This brutally shuts down the cascade. (Pathology Note: A genetic mutation called Factor V Leiden makes Factor V immune to destruction by APC, leading to severe hypercoagulability).
Mechanism: Directly blocks the Extrinsic pathway initiator. TFPI binds to and permanently inactivates the TF-VIIa-Xa complex, turning off the "initiator" tap.
B. Clot Dissolution (Fibrinolysis)
Once the injured vessel is repaired with new endothelial cells, the old fibrin scab must be dissolved to restore normal blood flow.
- The Key Enzyme - Plasmin: A powerful serine protease that chops up fibrin and fibrinogen, dismantling the clot.
- Activation: Plasmin floats in the blood in an inactive form called Plasminogen. It is converted into active Plasmin by Tissue Plasminogen Activator (t-PA) (released slowly by healed endothelial cells) or Urokinase (u-PA).
- Inhibitors of Fibrinolysis: The body uses PAI-1 (Plasminogen Activator Inhibitor-1) and Alpha-2-antiplasmin to ensure clots don't dissolve too early, which would cause re-bleeding.
- Breakdown Products: When Plasmin destroys cross-linked fibrin, it leaves behind distinctive trash molecules in the blood called Fibrin Degradation Products (FDPs), the most famous of which is the D-Dimer.
Clinical Application: "Clot Busters" (Thrombolytics)
If a patient arrives at the Emergency Department having an ischemic stroke (a clot blocking blood to the brain), doctors will inject synthetic recombinant t-PA (Alteplase). This drug forces massive, systemic conversion of Plasminogen to Plasmin, aggressively dissolving the clot in the brain and restoring blood flow. Because it destroys all clots, the major side effect is severe bleeding.
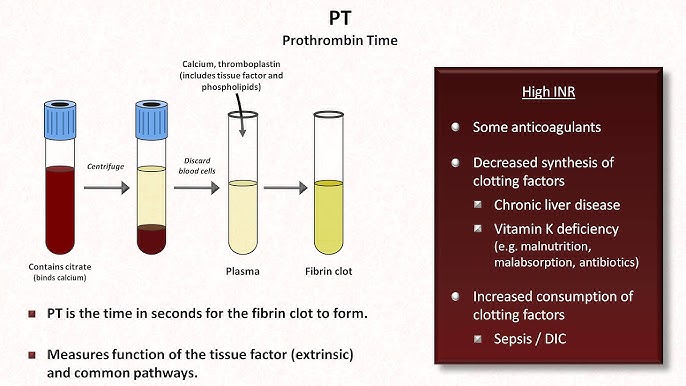
VI. Laboratory Tests for Hemostasis
Laboratory tests are vital for distinguishing whether a patient has a primary platelet defect or a secondary coagulation factor defect.
| Test Name | What it Measures | Normal Range | Clinical Significance & Abnormalities |
|---|---|---|---|
| Platelet Count | Total number of platelets in the blood. | 150,000 - 450,000 / µL | Low (Thrombocytopenia): Risk of mucosal bleeding. High (Thrombocytosis): Risk of thrombosis. |
| PFA-100 / Aggregometry | Platelet *function* (adhesion and aggregation). | Depends on assay | If platelet count is normal but the patient is bleeding, PFA-100 checks if the platelets are "lazy" or broken (e.g., von Willebrand Disease, Aspirin toxicity). |
| Prothrombin Time (PT) & INR | Measures the Extrinsic and Common Pathways. | PT: 10-14 seconds INR: 0.8 - 1.2 |
Mnemonic: "PeT" (PT = Extrinsic). Prolonged by deficiencies in VII, X, V, II, or Fibrinogen. Heavily used to monitor Warfarin therapy and liver failure. |
| Activated Partial Thromboplastin Time (aPTT) | Measures the Intrinsic and Common Pathways. | 25 - 35 seconds | Prolonged by deficiencies in XII, XI, IX, VIII (Hemophilia). Heavily used to monitor Heparin therapy. |
| D-Dimer | Measures the presence of degraded, cross-linked fibrin. | < 500 ng/mL | Highly Sensitive, Low Specificity. If elevated, it means the body is making and breaking clots (DVT, PE, DIC). If negative, you can confidently rule out a major blood clot. |
VII. Common Disorders of Hemostasis
Hemostasis disorders generally present in specific patterns. Primary disorders cause superficial bleeding (skin, mucous membranes), while secondary disorders cause deep tissue bleeding.
A. Primary Hemostasis Disorders (Platelet / Vessel Wall Defects)
Symptoms: Mucocutaneous bleeding (nosebleeds/epistaxis, bleeding gums, heavy menses), Petechiae (tiny pinpoint skin hemorrhages), and Purpura (larger purple skin bruises).
- Thrombocytopenia (Low Platelets):
- Decreased Production: Bone marrow failure, leukemia, chemotherapy, B12/folate deficiency.
- Increased Destruction: Immune Thrombocytopenic Purpura (ITP - antibodies destroy platelets), Thrombotic Thrombocytopenic Purpura (TTP - micro-clots shred platelets).
- Sequestration: Splenomegaly (an enlarged spleen acts as a sponge, trapping platelets).
- Platelet Function Disorders:
- Inherited: Glanzmann's thrombasthenia (missing GP IIb/IIIa, cannot aggregate), Bernard-Soulier syndrome (missing GP Ib, cannot adhere to vWF).
- Acquired: Aspirin/NSAID use, Uremia (severe kidney failure poisons platelets).
- Von Willebrand Disease (vWD): The most common inherited bleeding disorder. Patients lack functional vWF. Without vWF, platelets cannot stick to the wall (primary defect). Furthermore, vWF normally protects Factor VIII in the blood; without it, Factor VIII degrades rapidly (secondary defect).
Labs: Normal platelet count, abnormal PFA-100, prolonged aPTT (due to low Factor VIII).
B. Secondary Hemostasis Disorders (Coagulation Factor Defects)
Symptoms: Deep tissue bleeding, Hemarthroses (massive bleeding directly into joints causing swelling and pain), and large deep muscle hematomas.
- Hemophilia A (Factor VIII def.) & Hemophilia B (Factor IX def.): X-linked recessive genetic disorders almost exclusively affecting males. They cripple the Intrinsic pathway.
Labs: Normal PT, severely prolonged aPTT. - Vitamin K Deficiency: Seen in severe malnutrition, fat malabsorption (cystic fibrosis), or prolonged antibiotic use (kills gut flora that make Vit K). Affects Factors II, VII, IX, X.
Labs: Prolonged PT (very sensitive to Factor VII loss) and prolonged aPTT. - Liver Disease (Cirrhosis): The liver synthesizes almost all coagulation factors. Liver failure causes severe, multi-factorial bleeding tendencies.
Labs: Prolonged PT/INR, prolonged aPTT, low platelets (due to portal hypertension/splenomegaly).
Pathology Spotlight: Disseminated Intravascular Coagulation (DIC)
DIC is a catastrophic, paradoxical syndrome often triggered by severe sepsis, massive trauma, or obstetric emergencies. The massive systemic inflammation aggressively triggers the coagulation cascade everywhere at once, forming thousands of micro-clots throughout the body (causing organ failure and tissue necrosis). Because the body uses up ALL its platelets and coagulation factors making these micro-clots, the patient then begins to bleed uncontrollably from every orifice and IV site. It is a "consumption coagulopathy."
DIC Labs: Massively decreased Platelets, massively prolonged PT and aPTT, deeply low Fibrinogen, and a sky-high D-Dimer (due to the body frantically trying to dissolve all the micro-clots).
C. Thrombotic Disorders (Thrombophilia)
Conditions where the blood clots far too easily, leading to Deep Vein Thrombosis (DVT) or Pulmonary Embolisms (PE).
- Inherited Thrombophilias:
- Factor V Leiden: The most common genetic cause. A mutation makes Factor V highly resistant to being deactivated by Activated Protein C (APC). The cascade cannot be turned off.
- Prothrombin G20210A Mutation: Causes overproduction of prothrombin.
- Deficiencies of Natural Anticoagulants: Rare but severe genetic lack of Antithrombin, Protein C, or Protein S.
- Acquired Thrombophilias:
- Antiphospholipid Syndrome (APS): An autoimmune disorder where antibodies attack phospholipids, creating a highly pro-thrombotic state (often causing recurrent miscarriages). Lab paradox: APS causes clotting in the patient, but artificially prolongs the aPTT in a test tube.
- Heparin-Induced Thrombocytopenia (HIT): An immune reaction to Heparin drugs that causes widespread, deadly platelet activation and thrombosis.
- Other Causes: Cancer (malignancy secretes pro-coagulant mucins), Pregnancy (high estrogen increases clotting factors), and prolonged immobilization (surgery, long flights).
VIII. References and Further Reading
- Kumar, V., Abbas, A. K., & Aster, J. C. (2020). Robbins & Cotran Pathologic Basis of Disease (10th ed.). Elsevier. (Chapter on Hemodynamic Disorders, Thromboembolism, and Shock).
- Hall, J. E., & Hall, M. E. (2020). Guyton and Hall Textbook of Medical Physiology (14th ed.). Elsevier. (Chapter on Hemostasis and Blood Coagulation).
- Hoffman, R., Benz Jr, E. J., Silberstein, L. E., et al. (2017). Hematology: Basic Principles and Practice (7th ed.). Elsevier.
- Loscalzo, J., Fauci, A., Kasper, D., et al. (2022). Harrison's Principles of Internal Medicine (21st ed.). McGraw Hill. (Disorders of Hemostasis section).
Platelets and Hemostasis
Test your knowledge with these 25 questions.
Platelets and Hemostasis Quiz
Question 1/25
Quiz Complete!
Here are your results, .
Your Score
23/25
92%