
Erythrocytes, Hemoglobin, and Cellular Metabolism
By the conclusion of this exhaustive master guide, you will be deeply conversant with:
- The unique anatomical and structural adaptations of the Red Blood Cell (RBC).
- The intricate molecular architecture and functionality of Hemoglobin, including gas transport dynamics.
- The four specialized metabolic pathways of the erythrocyte that ensure its survival and function without organelles.
- The complete lifecycle of the erythrocyte: from Erythropoiesis in the bone marrow to Senescence and Destruction in the reticuloendothelial system.
Section I: Anatomy & Structural Physiology of the Erythrocyte
Red blood cells (RBCs), clinically referred to as erythrocytes, are arguably the most crucial cellular component of blood in terms of overall physiological function and homeostasis. Their primary mandate is the transport of life-sustaining oxygen from the pulmonary capillaries to the peripheral tissues, and the simultaneous transport of toxic carbon dioxide from the tissues back to the lungs for expiration. To execute this relentless, heavy-duty mechanical and chemical task, erythrocytes possess a highly specialized, stripped-down cellular structure.
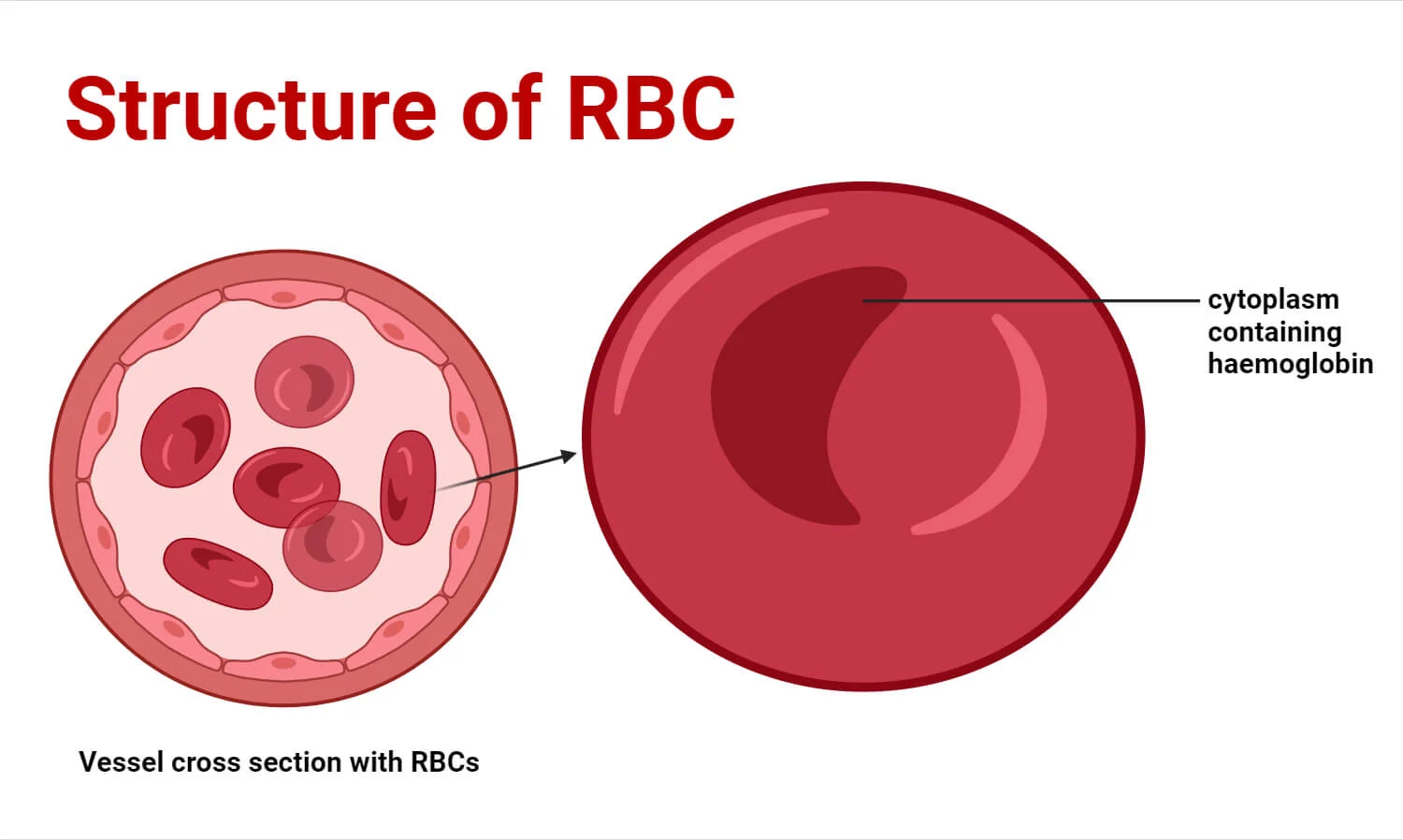
1. The Biconcave Disc Shape
Mature RBCs are incredibly flexible, anucleated entities. Their most visually defining characteristic is the biconcave disc shape—a flattened, doughnut-like structure with depressed, thinner centers on both sides. A normal erythrocyte measures approximately 7.5 µm in diameter, 2.0 µm in thickness at the extreme edges, and a mere 1.0 µm in the depressed center.
Functional Significance of the Biconcave Shape:
- Maximized Surface Area to Volume Ratio: This geometry provides a surface area approximately 30% greater than that of a sphere of the same volume. This massive surface area is absolutely critical for the rapid, efficient diffusion of O2 and CO2 gases across the cell membrane.
- Extreme Flexibility and Deformability: The human microvasculature contains capillaries that are often only 3 to 4 µm in diameter. An RBC (7.5 µm) must literally fold itself in half, parachute-style, to squeeze through these narrow endothelial gaps. The biconcave shape allows excess membrane slack, enabling this extreme deformation without rupturing the cell.
- Rouleaux Formation: The flattened nature of the discs allows RBCs to stack neatly on top of one another like a roll of coins (Rouleaux). This prevents turbulent flow and allows cells to glide smoothly through narrow venules and capillaries without jamming or causing microvascular occlusions.
2. Anucleated State & Lack of Organelles
During the final stages of maturation in the bone marrow, the erythrocyte commits the ultimate cellular sacrifice: it forcibly extrudes its nucleus and aggressively dismantles its mitochondria, endoplasmic reticulum, and Golgi apparatus.
Functional Significance:
- Maximized Hemoglobin Payload: By jettisoning bulky organelles, the erythrocyte frees up maximum internal volume to be packed almost entirely with hemoglobin. Approximately 97% of the cell's dry weight (non-water content) is pure hemoglobin.
- Zero Oxygen Consumption: Because they completely lack mitochondria, RBCs cannot perform oxidative phosphorylation. Consequently, they do not consume even a fraction of the precious oxygen they are transporting to the tissues. They operate entirely on anaerobic glycolysis.
- Limited Cellular Lifespan: The evolutionary trade-off for this extreme specialization is a short life. Lacking a nucleus and ribosomes, the mature RBC has absolutely no protein synthesis machinery. It cannot repair damaged enzymes or replace worn-out membrane lipids, rigidly limiting its lifespan to approximately 100 to 120 days.
3. The Specialized Plasma Membrane
The erythrocyte plasma membrane is a standard phospholipid bilayer, but it is heavily fortified on its intracellular surface by a dense, dynamic network of cytoskeletal proteins (predominantly Spectrin, Ankyrin, Actin, and Band 3).
Functional Significance:
- Maintaining Structural Integrity: The spectrin-actin cytoskeletal lattice acts like a flexible molecular scaffolding. It acts like springs, allowing the membrane to endure the massive, continuous shear stress of arterial blood pressure, and immediately snapping the cell back into its biconcave shape once it exits a narrow capillary.
- Antigen Presentation: The outer leaflet of the membrane is heavily studded with highly specific glycoproteins and glycolipids. These molecules act as structural identity markers, the most clinically significant being the ABO blood group antigens and the Rh (Rhesus) factor antigens, which dictate blood transfusion compatibility.
Hereditary Spherocytosis
To truly understand the importance of the spectrin cytoskeleton, we examine the genetic disease Hereditary Spherocytosis. Patients with this condition inherit a genetic mutation that causes a deficiency in spectrin or ankyrin proteins. Without this structural scaffolding, the RBC cannot maintain its biconcave shape and instead assumes a rigid, spherical shape (a spherocyte).
Because these spherocytes lack flexibility, they become trapped in the narrow cords of the spleen. The splenic macrophages view them as abnormal and aggressively devour them, leading to severe, chronic hemolytic anemia, massive splenomegaly (enlarged spleen), and jaundice.
Section II: Primary Functions of the Erythrocyte
1. Oxygen Transport
This is the prime directive of the RBC. Hemoglobin (Hb) binds reversibly to oxygen.
- In the Pulmonary Capillaries (Lungs): The environment boasts a high partial pressure of oxygen (PO2). Oxygen diffuses rapidly across the alveolar membrane into the RBC, loading onto the hemoglobin to form Oxyhemoglobin (HbO2). This oxygen-rich state gives arterial blood its characteristic bright, vibrant red appearance.
- In the Peripheral Tissues: The metabolic activity of the tissues depletes oxygen, creating an environment with a low PO2. The chemical bond between oxygen and hemoglobin weakens, causing the O2 to rapidly unload and diffuse into the surrounding tissue cells.
2. Carbon Dioxide Transport
As a byproduct of cellular metabolism, tissues generate massive amounts of toxic carbon dioxide (CO2). The RBC acts as a garbage truck, transporting CO2 back to the lungs via three distinct physiological methods:
The Bicarbonate Buffer System
The vast majority of CO2 diffuses into the RBC where an incredibly fast enzyme, Carbonic Anhydrase, catalyzes its reaction with water to form carbonic acid (H2CO3). This unstable acid instantly dissociates into a Hydrogen ion (H+) and a Bicarbonate ion (HCO3-).
The newly formed Bicarbonate is pumped out of the RBC into the blood plasma in exchange for a Chloride ion coming in (the Chloride Shift or Hamburger phenomenon). The bicarbonate safely travels in the plasma to the lungs.
Carbaminohemoglobin
A significant portion of CO2 binds directly to the terminal amino groups of the hemoglobin's globin protein chains (it does not bind to the iron where oxygen sits). This forms Carbaminohemoglobin (HbCO2). This reaction is highly reversible and heavily dependent on the local PCO2 levels in the blood.
Dissolved in Plasma
Because carbon dioxide is substantially more highly soluble in water than oxygen, a small but notable fraction simply remains dissolved as free gas within the fluid matrix of the blood plasma.
3. Systemic pH Regulation (Buffering)
Hemoglobin is not just a transporter; it is an exceptional biological buffer. When Carbonic Anhydrase converts CO2 into bicarbonate and a free, highly acidic Hydrogen ion (H+), this threatens to catastrophically drop the blood pH. However, Deoxyhemoglobin (hemoglobin that has just dropped off its oxygen) has an incredibly high chemical affinity for H+ ions. It acts like a sponge, soaking up the free H+ ions, thereby preventing intracellular acidosis and maintaining strict systemic blood pH within the narrow, life-sustaining physiological window of 7.35 to 7.45.
Section III: Molecular Architecture and Function of Hemoglobin
Hemoglobin (Hb) is the specialized, complex globular protein completely responsible for the erythrocyte's gas-carrying capacity. It boasts a complex quaternary protein structure that is evolutionarily perfected for its role in gas exchange.
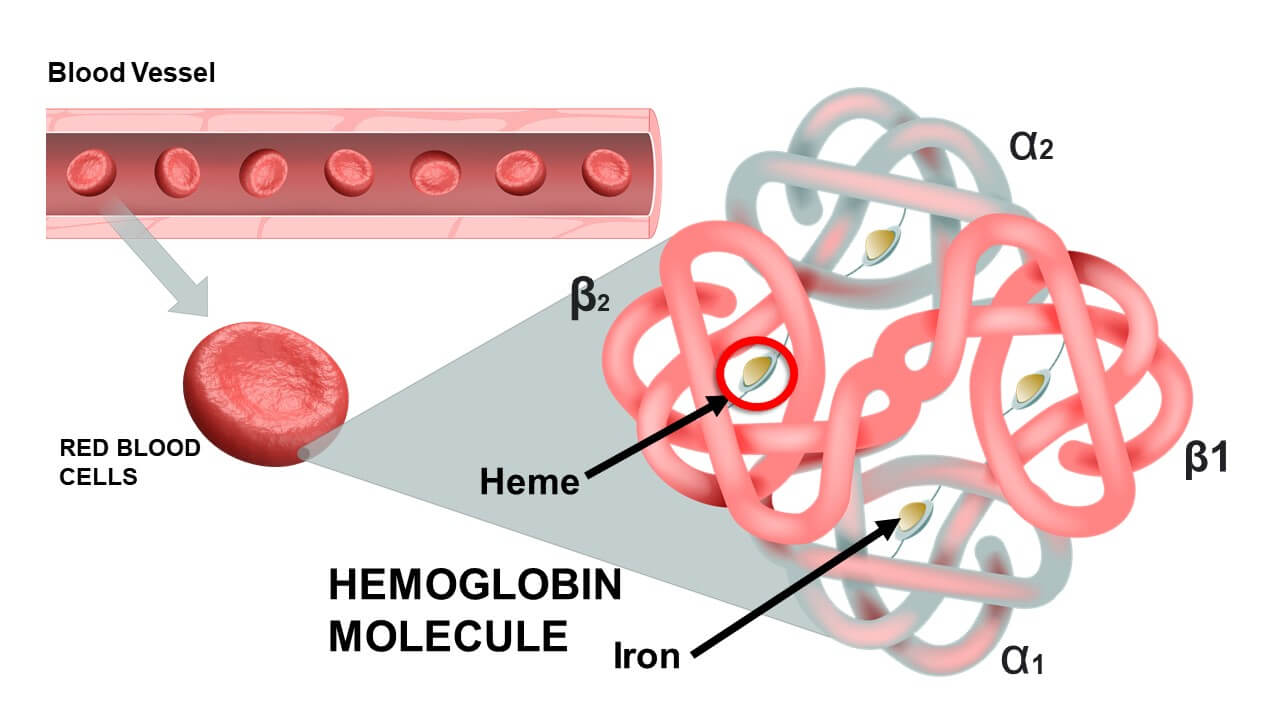
1. The Structure of Hemoglobin
- Four Polypeptide Chains (Globins): A single, complete hemoglobin molecule is constructed from four highly folded protein subunits. In healthy adults, the overwhelmingly dominant type (HbA) consists of two Alpha (α) chains and two Beta (β) chains. Each globin chain features a highly specific amino acid sequence folded into an intricate 3D structure.
- Four Heme Groups: Embedded deep within the folds of each of the four globin chains is a non-protein, iron-containing prosthetic group called a Heme group. Thus, 1 complete Hemoglobin molecule = 4 distinct Heme groups.
- The Porphyrin Ring: Specifically known as Protoporphyrin IX, this is a large, flat, complex organic ring structure.
- The Iron Ion (Fe2+): Suspended perfectly in the dead center of the porphyrin ring is a single iron ion. Critical Physiology: This iron MUST be maintained in the Ferrous (Fe2+) state. If it is oxidized to the Ferric (Fe3+) state, it forms Methemoglobin, which is utterly incapable of binding oxygen.
2. Advanced Functional Dynamics of Hemoglobin
A. Cooperative Binding and the Sigmoidal Curve
Hemoglobin does not bind oxygen passively; it utilizes a highly dynamic biochemical phenomenon known as Cooperative Binding (allosteric modification). In the deoxygenated state, hemoglobin exists in a tight, rigid conformation known as the T-State (Tense state), which has a low affinity for oxygen.
When the very first O2 molecule forces its way in and binds to one of the four heme groups, it breaks several salt bridges. This causes a dramatic conformational (shape) change in the entire hemoglobin molecule, snapping it into the R-State (Relaxed state). This shape change massively increases the chemical affinity of the remaining three empty heme groups for oxygen. As each oxygen binds, the next one binds even faster.
This cooperative action results in the classic S-shaped (sigmoidal) Oxygen-Hemoglobin Dissociation Curve. It ensures that Hb loads up completely in the lungs and unloads rapidly and massively the moment it detects low oxygen in the peripheral tissues.
B. The Bohr and Haldane Effects
- The Bohr Effect (Tissue Level): Increased CO2 and increased H+ (acidity) in actively working tissues physically forces hemoglobin to release its oxygen more readily.
- The Haldane Effect (Lung Level): Conversely, the high concentration of Oxygen in the lungs forces hemoglobin to aggressively dump its loaded CO2 and H+ ions so they can be exhaled. Oxygenation of blood in the lungs naturally displaces carbon dioxide from hemoglobin.
3. Variants and Types of Hemoglobin
The classification of hemoglobin types is based entirely on the specific composition of their polypeptide globin chains.
| Hemoglobin Type | Globin Chain Structure | Prevalence & Clinical Significance |
|---|---|---|
| Hemoglobin A (HbA) Adult |
2 Alpha (α) + 2 Beta (β) chains. (α2β2) |
The overwhelmingly dominant form in healthy adults, accounting for 95% to 98% of all circulating hemoglobin. |
| Hemoglobin A2 (HbA2) Minor Adult |
2 Alpha (α) + 2 Delta (δ) chains. (α2δ2) |
A normal, minor variant comprising roughly 1.5% to 3.5% of adult hemoglobin. |
| Hemoglobin F (HbF) Fetal |
2 Alpha (α) + 2 Gamma (γ) chains. (α2γ2) |
The primary hemoglobin of the developing fetus. Crucial Physiology: HbF does not bind 2,3-BPG well. Therefore, it has a vastly higher affinity for oxygen than adult HbA. This allows the fetal blood to literally "steal" oxygen across the placenta from the mother's red blood cells. |
Hemoglobinopathies: Sickle Cell and Thalassemia
Genetic defects in the DNA instructions for globin chains lead to devastating hemoglobinopathies.
- Sickle Cell Anemia: Caused by a single point mutation (Glutamic acid replaced by Valine) in the Beta-globin chain, creating abnormal HbS. Under low oxygen conditions, HbS molecules polymerize (clump together) into long, rigid crystalline rods. This physically distorts the RBC into a sharp, rigid "sickle" shape. These sickle cells rupture easily (hemolysis) and jam together to block small blood vessels, causing excruciating pain crises, organ ischemia, and tissue infarction.
- Thalassemias: Unlike Sickle Cell (where the protein is built wrong), Thalassemias occur when the bone marrow simply doesn't build enough of the protein. Alpha-Thalassemia is a decreased synthesis of Alpha chains; Beta-Thalassemia is a decreased synthesis of Beta chains. This results in tiny, pale RBCs (microcytic, hypochromic anemia) and severe oxygen starvation.
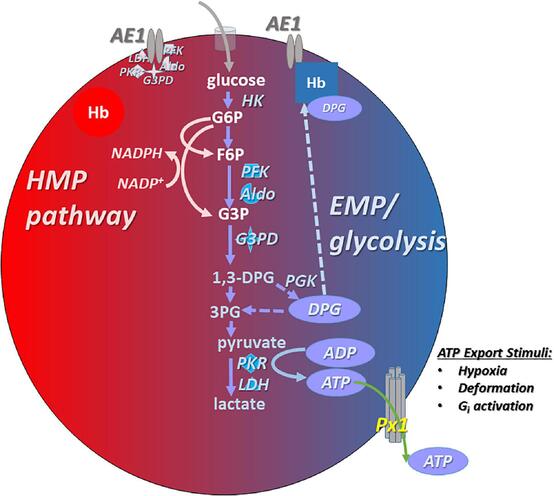
Section IV: Metabolic Pathways of the Erythrocyte
Because the mature red blood cell purposefully lacks a nucleus, mitochondria, and other complex organelles, it relies on a highly simplified, stripped-down, yet exquisitely specialized metabolic machinery. Its entire biochemistry is designed to do exactly two things: keep the cell alive and keep the hemoglobin functional.
1. Primary Energy Production: Anaerobic Glycolysis (Emden-Meyerhof Pathway)
The Dilemma: Lacking mitochondria, RBCs cannot perform oxidative phosphorylation or utilize the Krebs cycle. If they used the oxygen they transport for energy, they would steal it from the brain and heart.
The Solution: Glycolysis is the sole, exclusive pathway for ATP generation in the mature RBC. This pathway consumes glucose directly from the blood plasma, breaking it down into pyruvate, yielding a meager but essential net gain of 2 ATP molecules per glucose molecule.
The End Product: Without mitochondria to process the pyruvate, an enzyme called Lactate Dehydrogenase instantly converts the pyruvate into Lactate (lactic acid). This lactate diffuses out of the RBC into the plasma, travels to the liver, and is recycled back into glucose via gluconeogenesis (the Cori Cycle).
What does the RBC do with this ATP?
- Fueling Ion Pumps: ATP exclusively powers the membrane-bound Na+/K+-ATPase pumps. These pumps exhaustively eject sodium out of the cell to prevent water from rushing in via osmosis. If ATP fails, the cell swells and explodes (osmotic hemolysis).
- Cytoskeletal Maintenance: ATP is required to phosphorylate and maintain the elastic tension of the spectrin-actin lattice, keeping the cell biconcave.
2. The Hexose Monophosphate (HMP) Shunt / Pentose Phosphate Pathway
While this side pathway produces exactly zero ATP, it is the only thing keeping the RBC from oxidizing and rusting from the inside out.
- The Product: About 10% of glucose is shunted into this pathway to generate NADPH (Nicotinamide Adenine Dinucleotide Phosphate).
- The Mechanism of Protection: Hemoglobin continuously generates highly toxic Reactive Oxygen Species (ROS), like Hydrogen Peroxide (H2O2). The RBC defends itself using an antioxidant protein called Glutathione (GSH). The enzyme Glutathione Peroxidase uses GSH to neutralize the dangerous H2O2 into harmless water. However, this process "uses up" the Glutathione, oxidizing it into GSSG. The NADPH generated by the HMP shunt is strictly used by the enzyme Glutathione Reductase to recycle the GSSG back into functional, active GSH.
Clinical Correlate: G6PD Deficiency
Glucose-6-Phosphate Dehydrogenase (G6PD) is the rate-limiting enzyme that starts the HMP Shunt. It is one of the most common genetic enzyme deficiencies worldwide. Patients with G6PD deficiency cannot produce enough NADPH.
Under normal conditions, they are fine. But if they are exposed to massive oxidative stress—such as eating Fava Beans, taking Sulfa antibiotics, using specific Antimalarial drugs (Primaquine), or fighting a severe bacterial infection—the generated H2O2 is not neutralized. The toxic ROS immediately destroy the hemoglobin, precipitating it into hard, destructive chunks called Heinz Bodies. As these RBCs pass through the spleen, macrophages take a literal bite out of the membrane to remove the Heinz body (creating Bite Cells), leading to massive, rapid hemolytic anemia.
3. The Rapoport-Luebering Shunt (2,3-Bisphosphoglycerate Pathway)
This is a unique metabolic side-branch of glycolysis found almost exclusively in erythrocytes.
- The Purpose: Instead of proceeding down standard glycolysis to make ATP, an enzyme called Bisphosphoglycerate mutase converts an intermediate into 2,3-Bisphosphoglycerate (2,3-BPG) (also historically called 2,3-DPG).
- The Mechanism: 2,3-BPG acts as a powerful allosteric modulator. It physically wedges itself into the center of the deoxygenated hemoglobin molecule. By binding there, it forces the hemoglobin to lock into the rigid "T-State", vastly decreasing the hemoglobin's affinity for oxygen.
- Clinical Significance: This forces the RBC to drop more oxygen into the tissues. If a person climbs a high mountain (hypoxia) or suffers from chronic lung disease (COPD), their RBCs massively ramp up the Rapoport-Luebering shunt to produce excessive 2,3-BPG, ensuring their starving tissues get maximum oxygen delivery. (This physically causes a "Right Shift" on the oxygen-hemoglobin dissociation curve).
4. The Methemoglobin Reductase Pathway
Oxygen naturally oxidizes the functional Ferrous iron (Fe2+) in hemoglobin into non-functional Ferric iron (Fe3+) at a slow, continuous rate of about 3% per day. This useless form is called Methemoglobin.
- The Defense: The RBC utilizes the enzyme Methemoglobin Reductase (also called Cytochrome b5 reductase). This enzyme uses NADH (harvested from standard glycolysis) to efficiently reduce the Ferric iron (Fe3+) straight back into functional Ferrous iron (Fe2+).
- Clinical Correlate: If an individual is exposed to excessive oxidizing toxins (like nitrites, local anesthetics like benzocaine, or specific antibiotics), the pathway is overwhelmed. The patient develops Methemoglobinemia. Their blood turns a thick, dark chocolate-brown color, and they exhibit profound "chocolate cyanosis" (blue/gray skin) because their blood can no longer carry oxygen. The emergency antidote is an IV infusion of Methylene Blue, which artificially accelerates the reductase enzyme to save the patient.
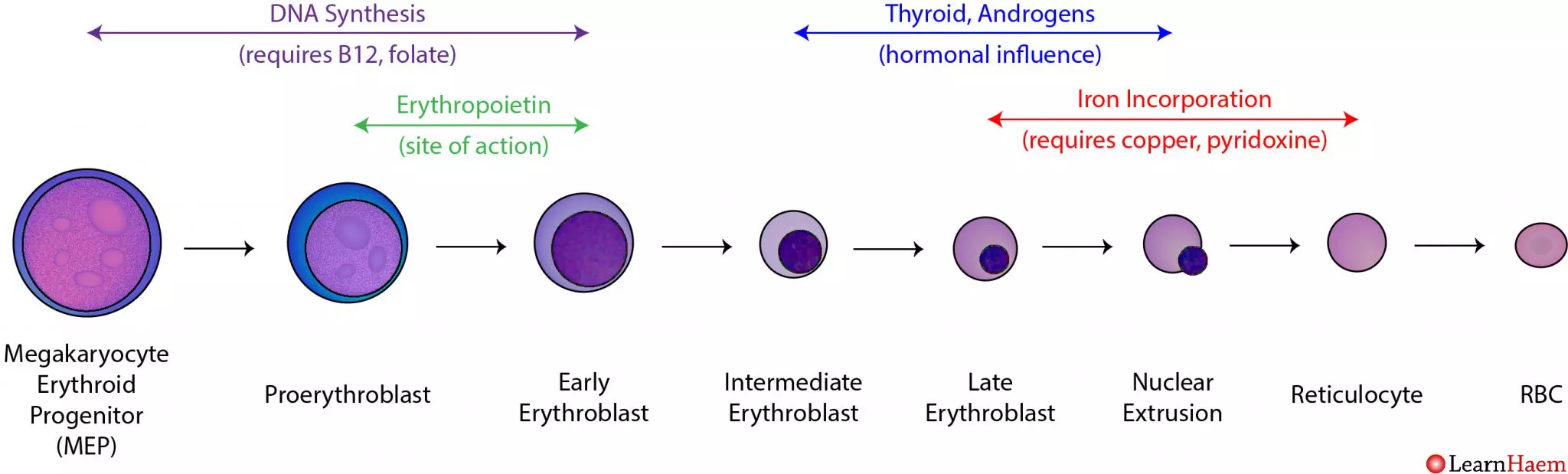
Section V: Erythropoiesis: The Birth of the Red Blood Cell
The lifespan of an RBC is short. To compensate for the loss of millions of cells per second, the body utilizes Erythropoiesis—the highly orchestrated, dynamic production of new red blood cells originating in the red bone marrow.
1. The Hypoxic Stimulus and Erythropoietin (EPO)
The bone marrow does not guess how many RBCs to make; it waits for a precise hormonal order.
- The Sensor: The peritubular interstitial cells of the Kidneys are incredibly sensitive to oxygen tension. If blood oxygen levels drop (due to high altitude, blood loss, or pulmonary disease), the kidneys detect the hypoxia.
- The Hormone: In response to hypoxia, the kidneys synthesize and secrete massive amounts of the glycoprotein hormone Erythropoietin (EPO) into the bloodstream.
- The Target: EPO travels directly to the red bone marrow and binds to specific receptors on committed hematopoietic stem cells, slamming the accelerator on cellular proliferation, accelerating maturation, and triggering hemoglobin synthesis.
Chronic Kidney Disease & Anemia
Patients suffering from end-stage renal disease (kidney failure) universally develop profound, chronic anemia. Why? Because their destroyed, fibrotic kidneys can no longer produce or secrete Erythropoietin. Without EPO, the bone marrow simply halts RBC production. Modern medicine treats this by injecting these patients with synthetic recombinant human Erythropoietin (rHuEPO) to forcefully restart the bone marrow.
2. The Sequential Stages of Erythropoiesis
The transformation from a generic stem cell to a highly specialized anucleated disc takes about 5 to 7 days, following an exact lineage:
- Hematopoietic Stem Cell (HSC): The multipotent granddaddy of all blood cells.
- Common Myeloid Progenitor (CMP): The cell commits to the myeloid line (excluding lymphocytes).
- Proerythroblast (Pronormoblast): The first committed, recognizable red cell precursor. It is massive, with a huge nucleus and dark blue (basophilic) cytoplasm packed with millions of ribosomes preparing to build protein.
- Basophilic Erythroblast: Rapid cellular division occurs. The nucleoli disappear.
- Polychromatic Erythroblast: The critical turning point. The cell begins actively synthesizing large amounts of red-staining Hemoglobin. The mixture of blue ribosomes and red hemoglobin causes the cytoplasm to stain a murky purple/gray (polychromatic).
- Orthochromatic Erythroblast (Normoblast): The cell is now fully packed with hemoglobin, staining a vibrant pink (eosinophilic). The nucleus ceases all function, condensing into a tight, dark, dead mass (pyknosis) before being forcefully ejected from the cell.
- Reticulocyte: The cell is now anucleated but still contains a residual, web-like net (reticulum) of ribosomal RNA. Reticulocytes are released from the bone marrow into the peripheral bloodstream. They circulate for exactly 1 to 2 days before the spleen plucks out the remaining RNA, finalizing their maturation.
Clinical Note: Measuring the blood Reticulocyte Count provides a direct, real-time window into bone marrow function. A high count means the marrow is working overtime (e.g., recovering from bleeding); a low count means the marrow is failing. - Mature Erythrocyte: The final, perfect biconcave disc ready for 120 days of service.
3. Absolute Nutritional Requirements
Erythropoiesis demands extreme amounts of raw building materials.
- Iron: The non-negotiable core of the heme group. It is absorbed in the duodenum, bound to the transport protein Transferrin in the blood, and delivered to the marrow. Excess is stored as Ferritin in the liver. A lack of iron results in Iron Deficiency Anemia (small, pale, empty RBCs).
- Vitamin B12 (Cobalamin) and Folate (Folic Acid): These two vitamins are absolutely vital for rapid DNA synthesis and cellular division. If either is missing, the DNA cannot replicate fast enough to divide. The cell cytoplasm continues to grow and fill with hemoglobin, but the nucleus lags behind (nuclear-cytoplasmic asynchrony). The result is the production of massive, fragile, dysfunctional red blood cells—a condition known as Megaloblastic Anemia (or Pernicious Anemia if driven by an autoimmune lack of Intrinsic Factor needed for B12 absorption).
- Amino Acids: Massive amounts of dietary protein are required to synthesize the millions of globin polypeptide chains.
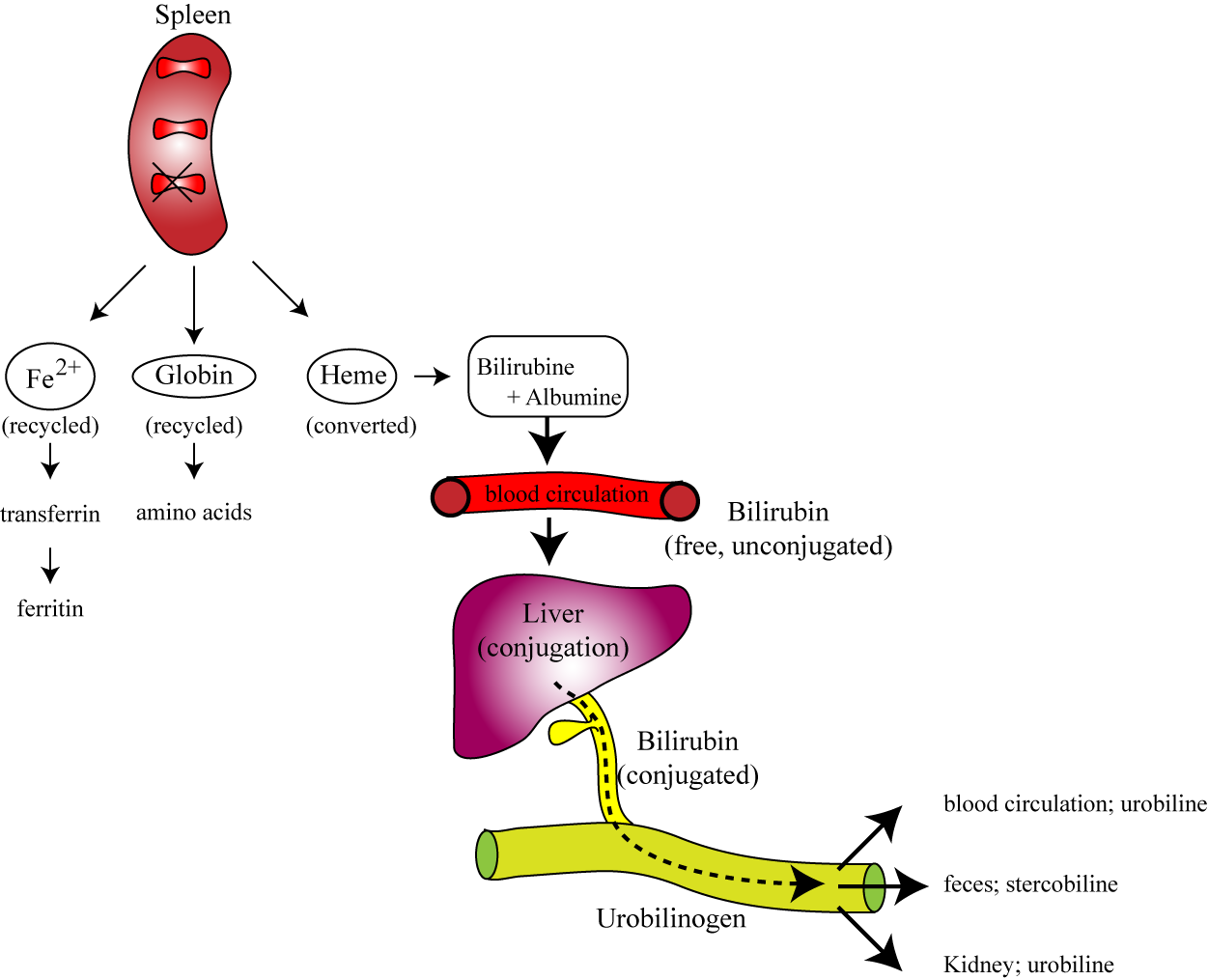
Section VI: Erythrocyte Senescence and Destruction
After a grueling 120-day journey, covering over 300 miles of vascular pathways, the RBC reaches the end of its life (senescence). Its enzymes fail, ATP is depleted, and the spectrin membrane becomes dangerously stiff, rigid, and fragile.
1. The Role of the Reticuloendothelial System (RES)
The vast majority (90%) of RBC destruction occurs smoothly and silently via Extravascular Hemolysis. The spleen acts as the ultimate quality-control filter. Old, rigid RBCs cannot squeeze through the tiny, hostile sinusoidal slits of the splenic red pulp. They become trapped. Waiting resident macrophages immediately recognize the damaged membrane proteins and ruthlessly phagocytize (devour) the aged RBCs. (The Kupffer cells of the liver and bone marrow macrophages assist in this process).
2. The Biochemical Breakdown and Recycling of Hemoglobin
Nothing goes to waste. The macrophage acts as a recycling center:
- Globin Chains: The massive protein structures are brutally catabolized by proteases into their individual constituent amino acids. These amino acids are dumped back into the blood plasma to be reused for general cellular protein synthesis or new erythropoiesis.
- The Heme Group: The heme ring requires delicate dismantling:
- Iron Extraction: The valuable Iron (Fe2+) is meticulously salvaged from the center of the ring. It is pushed out of the macrophage, binds to Transferrin in the blood, and is securely transported back to the bone marrow to build new hemoglobin, or stored safely as Ferritin.
- Porphyrin Ring Degradation: The empty organic ring is toxic and must be removed. The macrophage enzyme Heme Oxygenase rips the ring open, creating a green pigment called Biliverdin (with the release of a tiny amount of Carbon Monoxide gas).
- Unconjugated Bilirubin: Biliverdin is rapidly reduced by Biliverdin Reductase into a yellow/orange toxic waste pigment called Unconjugated (Indirect) Bilirubin. Because this molecule is highly lipid-soluble and utterly water-insoluble, it must bind tightly to the plasma protein Albumin to safely travel through the bloodstream to the liver.
- Hepatic Conjugation: Inside the liver hepatocytes, the enzyme UDP-glucuronosyltransferase (UGT) forces glucuronic acid onto the bilirubin. This transforms it into Conjugated (Direct) Bilirubin, making it highly water-soluble. The liver excretes this safe, water-soluble conjugated bilirubin into the bile duct, draining it into the small intestines to help emulsify dietary fats.
- Intestinal Excretion: In the large intestine, normal gut flora (bacteria) metabolize the conjugated bilirubin into Urobilinogen. A tiny fraction is reabsorbed into the blood and filtered by the kidneys, oxidizing into Urobilin (giving urine its characteristic yellow color). The vast majority remains in the gut, undergoing oxidation into Stercobilin, which is exclusively responsible for giving human feces its characteristic brown color.
Clinical Pathology: Jaundice (Icterus)
If any step in the RBC destruction and bilirubin clearance pathway fails, bilirubin accumulates massively in the blood (hyperbilirubinemia). Because bilirubin is yellow, it deposits in the skin and the sclera (white parts) of the eyes, causing intense yellowing known as Jaundice. We classify this diagnostically:
- Pre-hepatic Jaundice: Massive RBC destruction (e.g., Sickle cell crisis, Malaria, G6PD attack). The spleen crushes so many RBCs that it creates more unconjugated bilirubin than the liver can possibly handle.
- Hepatic Jaundice: The liver itself is sick (e.g., Viral Hepatitis, Cirrhosis) and simply lacks the cellular machinery to conjugate the normal daily load of bilirubin.
- Post-hepatic (Obstructive) Jaundice: The liver works perfectly and conjugates the bilirubin, but a physical blockage (e.g., a massive gallstone blocking the bile duct or pancreatic cancer) prevents the bile from draining into the intestines. The conjugated bilirubin backs up into the blood, causing dark urine and remarkably pale, clay-colored feces (because no stercobilin is made).
Section VII: List of References
- Hall, J. E., & Guyton, A. C. (2015). Guyton and Hall Textbook of Medical Physiology (13th ed.). Philadelphia, PA: Elsevier Saunders. (Chapters detailing Erythrocytes, Anemia, and Polycythemia).
- Kumar, V., Abbas, A. K., & Aster, J. C. (2020). Robbins & Cotran Pathologic Basis of Disease (10th ed.). Philadelphia, PA: Elsevier. (Sections on Hemodynamic Disorders, Hemoglobinopathies, and Jaundice).
- Hoffbrand, A. V., & Steensma, D. P. (2019). Hoffbrand's Essential Haematology (8th ed.). Wiley-Blackwell. (Comprehensive chapters on Erythropoiesis, RBC metabolism, and Hemolytic Anemias).
- Ferrier, D. R. (2017). Lippincott Illustrated Reviews: Biochemistry (7th ed.). Wolters Kluwer. (In-depth analysis of the Pentose Phosphate Pathway, Glycolysis, and Hemoglobin Structure/Function).
- Mescher, A. L. (2018). Junqueira's Basic Histology: Text and Atlas (15th ed.). McGraw-Hill Education. (Ultrastructural details of the erythrocyte cytoskeleton and bone marrow architecture).
Red Blood Cells Quiz
Test your knowledge with these 35 questions.
Red Blood Cells Quiz
Question 1/35
Quiz Complete!
Here are your results, .
Your Score
33/35
94%