Introduction to Basic Physiology & The Cell
Physiology is the rigorous scientific discipline dedicated to studying the functional activities and intricate mechanisms within a biological body. For example: why can the heart automatically beat, and how do electrical impulses coordinate this action? The word Physiology is derived from two ancient Greek words: physis, meaning "nature"; and logos, meaning "study". Thus, it is the study of the nature of living things.
1. Physiology Involves Process and Function
Words, names, and terms are of paramount importance in any scientific discipline because they carry highly precise meanings. Knowing and deeply understanding the relationships and etymological roots of these words will help tremendously in remembering and comprehending the vast amounts of information in a much deeper, more permanent way. This foundational knowledge will stay with you long after the course is over, enabling you to recognize important elements in other medical disciplines when you connect them to their deeper meanings.
Defining Physiology
The etymology (word origin) of the term Physiology traces back to 1560’s French, descending directly from the Latin physiologia, meaning “The study and description of natural objects, natural philosophy". This, in turn, is derived from the Greek physios meaning "nature, natural, physical"; and logia meaning "study". This gives us the fullest, most accurate meaning of Physiology as the "Science of the normal function of living things". When studying physiology, it is absolutely imperative that we also deeply understand the basic anatomy involved, as anatomy (structure) and physiology (function) are two sides of the same coin and go hand in hand.
Defining Anatomy
The etymology of the term Anatomy originates from Late 1300’s terminology in both Latin (anatomia) and Greek (anatome). These ancient words are derived from ana which means "up"; and tomos (or temnein) which means "to cut". Together this translates to "a cutting up", which is clearly referencing the act of physical dissection! In general clinical terms, anatomy is considered the “Study or knowledge of the structure (form) and organization of the human body“. While courses and textbooks for anatomy and physiology are often separated for convenience, they are inextricably connected to each other. A structure is designed specifically to support its function, and a function can only be carried out by an appropriately designed structure.
2. Etymology for the Language of Physiology
Another incredibly useful concept related to the importance of words in physiology (and anatomy) is mastering the etymology (origin of the word) of the vast array of scientific terms used in the healthcare field. Since the overwhelming majority of these words are derived from Latin and Greek, it is incredibly helpful to know the origins and ‘translations’ of these prefixes, roots, and suffixes. Becoming highly aware of the origins of words will greatly help students to:
- Understand exactly what a complex medical term means without memorizing it blindly.
- Assist you in instantly predicting what a brand-new, unseen term means when you first encounter it in a clinical setting.
Example 1: Hypertonic
The solution is hypertonic. Hyper means "above normal" or "excessive", and tonic refers to "strength" or "tension". Therefore, the solution is strong, excessively concentrated, and has a high osmotic pressure compared to another fluid.
Example 2: Hypoglycemia
The patient has hypoglycemia. Hypo is the direct opposite of hyper and means "below normal" or "deficient". The glyc portion refers to glucose (a type of sugar), and emia means "condition of the blood". Therefore, this statement translates perfectly to: the person has abnormally low blood sugar levels.
Example 3: Hyponatremia
A marathon runner presents with hyponatremia. Hypo still means "below normal". The natr portion comes from natrium, which is the Latin word for sodium (this is exactly why the chemical symbol for sodium on the periodic table is Na), and emia still means "blood". Therefore, this diagnosis means the person has dangerously low sodium levels in their blood.
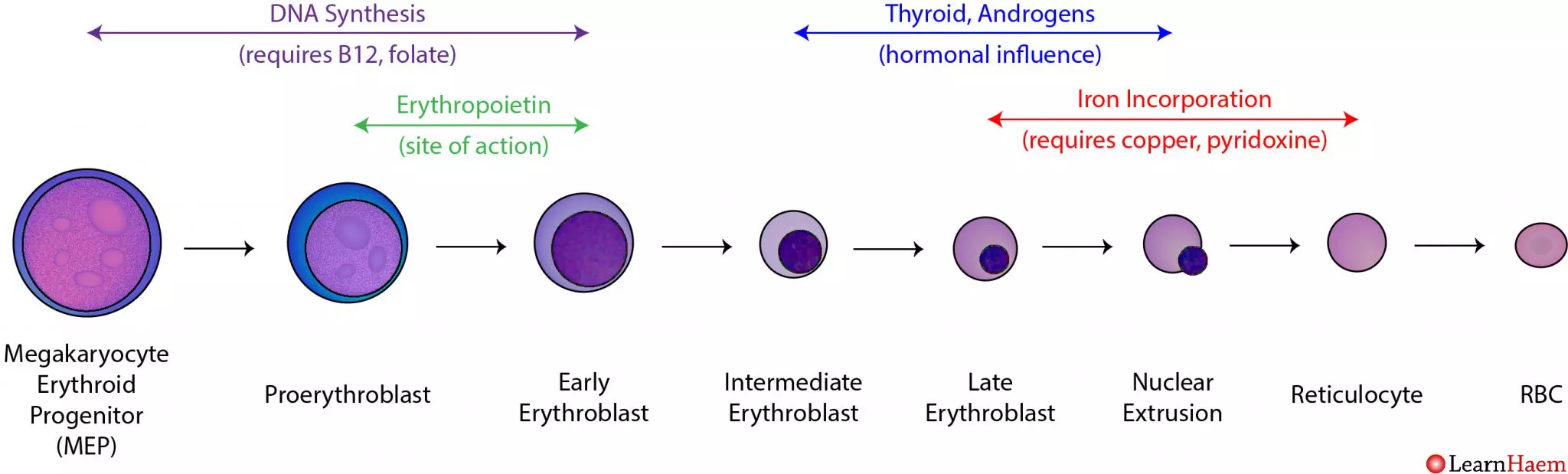
Extra Example: Erythropoiesis
The bone marrow undergoes erythropoiesis. Erythro means "red", and poiesis means "to make" or "formation". Thus, this term describes the physiological process of producing new red blood cells.
Along the way in this physiology course, we will encounter many of these terms that, once we know the origin and meaning of, will help us figure out newer terms with absolute ease and familiarity. Anyone who has taken a medical terminology course will know the immense value of understanding the meaning of roots, prefixes, and suffixes.
Interactive Exercise: Decode the Term
Diagnosis: Pancytopenia. (Hint: there are 3 distinct root terms here: pan, cyto, and penia).
The Detailed Answer:
• Pan means "all" or "every" (like a pandemic, which affects all people).
• Cyto means "cell".
• Penia means "deficiency", "lack of", or "poverty".
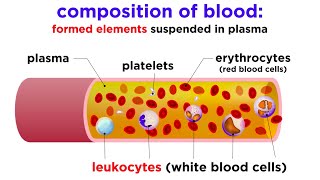
Translation: A deficiency of ALL cellular components in the blood. A patient with pancytopenia has dangerously low levels of red blood cells (anemia), white blood cells (leukopenia), and platelets (thrombocytopenia), usually due to complete bone marrow failure.
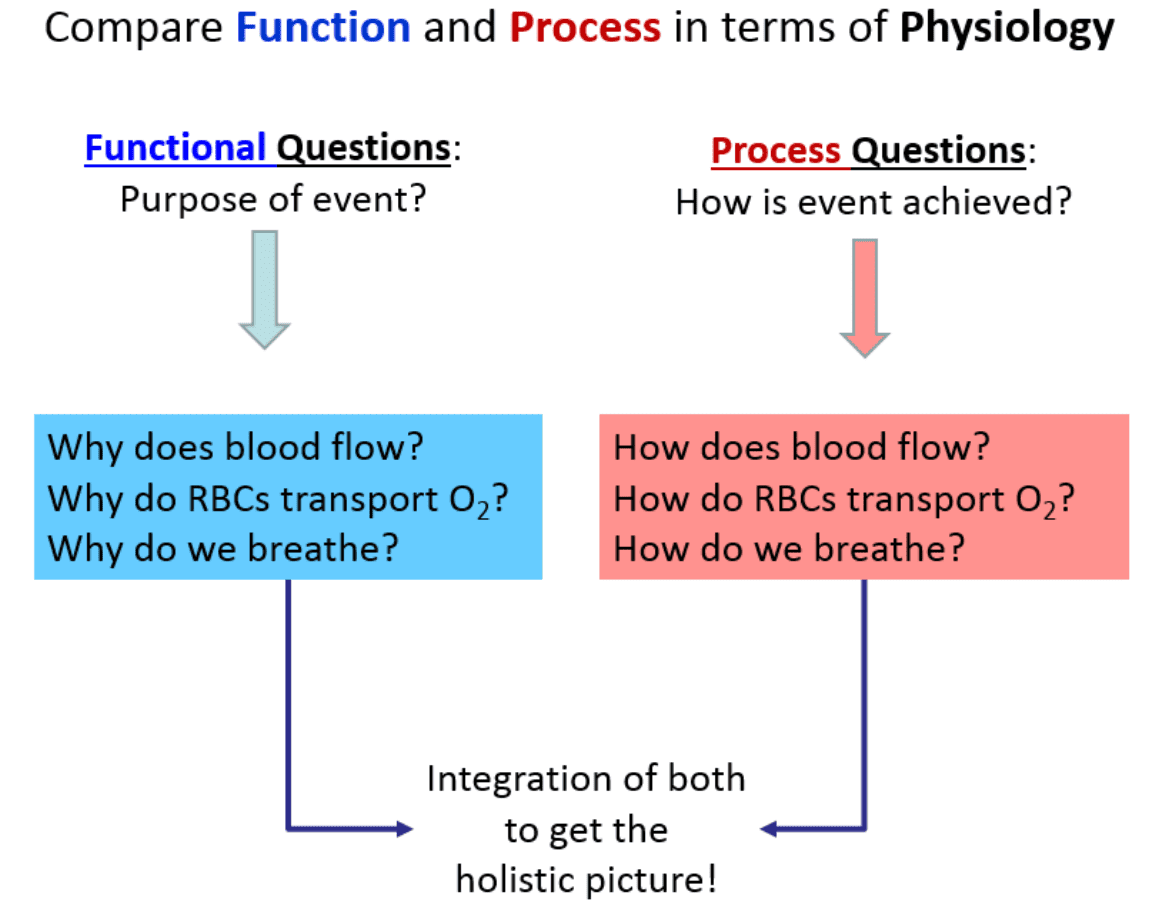
3. Compare Function and Process in Human Physiology
As we look to understand the central themes of physiology, an important concept is learning exactly how to ask questions about what’s occurring in the human body. In general, there are two basic, distinct approaches to physiology: 1) We can ask Functional Questions; and 2) We can ask Process Questions.
1. Functional Questions (The "Why")
These are strictly related to Why something occurs. They seek to understand the ultimate purpose or the evolutionary advantage of a mechanism. For example, what is the survival purpose of the heart beating? These can often be answered from a "big picture" perspective without requiring microscopic detail.
- Q: Why does blood flow?
A: To continuously transport vital nutrients, hormones, and gases (oxygen) around the body to sustain tissues, and to carry away toxic metabolic wastes (carbon dioxide, urea) to the excretory organs.
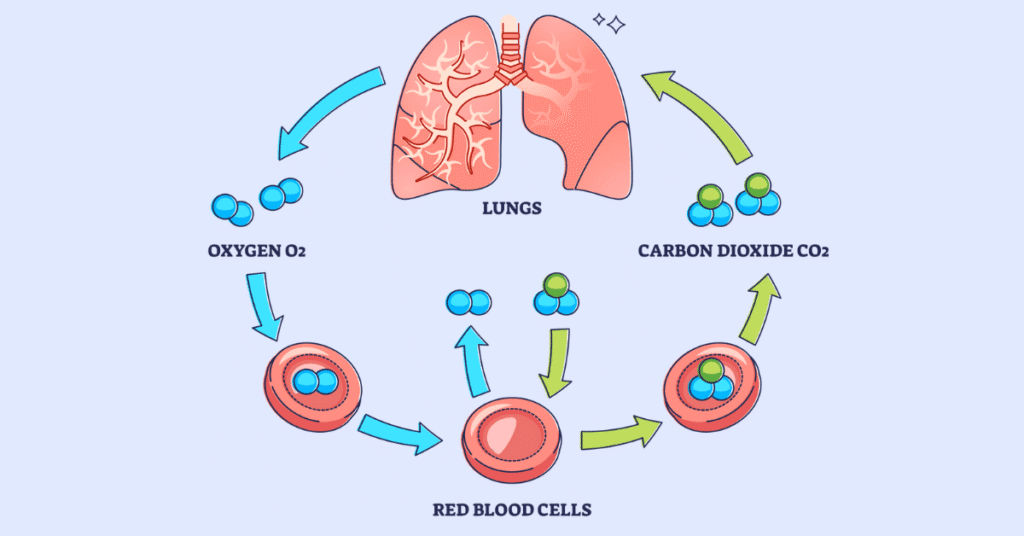
- Q: Why do Red Blood Cells (RBCs) transport O₂?
A: To efficiently deliver oxygen to all the body tissues that desperately need it to perform aerobic cellular respiration and generate ATP (energy).
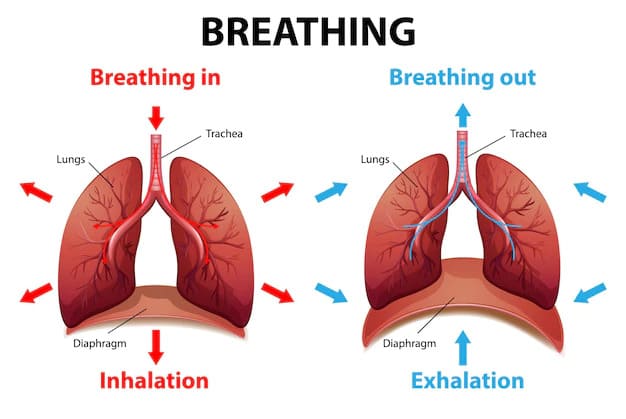
- Q: Why do we breathe?
A: To extract essential oxygen (O₂) from the inhaled atmospheric air and to continuously release volatile carbon dioxide (CO₂) back out of the body, thus preventing fatal acid build-up in the blood.
2. Process Questions (The "How")
These are related to How something physically and chemically occurs. For example, how does the heart actually manage to beat? Often these issues must be answered in an extensively detailed, step-by-step, mechanistic manner.
- Q: How does blood flow?
A: The muscular ventricles of the heart contract (systole) to generate a massive high-pressure wave. Blood flows strictly down this pressure gradient, moving from the high-pressure aorta through the progressively lower-pressure arteries, capillaries, and veins, finally returning to the heart aided by skeletal muscle pumps and tissue fluid pressures.
- Q: How do RBCs transport O₂?
A: Inside the red blood cells, the iron-containing heme portion of the hemoglobin molecule undergoes conformational changes. It exhibits a high chemical affinity for O₂ when the surrounding partial pressure of O₂ is high (such as in the pulmonary capillaries of the lungs, forcing O₂ to bind). Conversely, it exhibits a low affinity for O₂ when the surrounding partial pressure for O₂ is low (such as in metabolically active, oxygen-depleted muscle tissue, forcing the hemoglobin to drop the O₂ where it is needed).
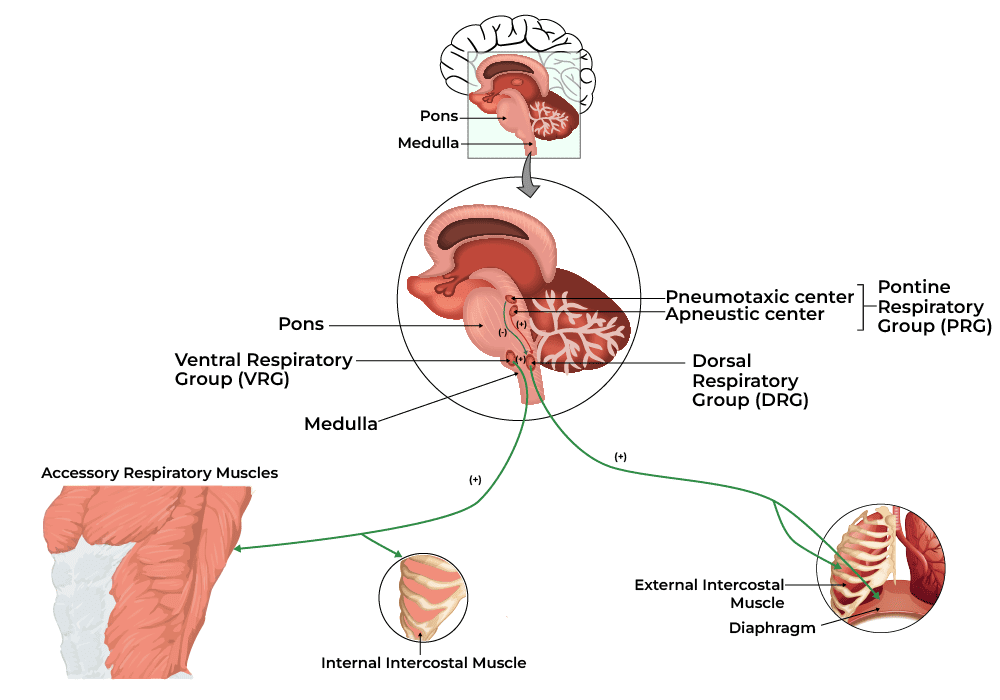
- Q: How do we breathe?
A: The diaphragm and external intercostal muscles (skeletal muscles of respiration) contract, pulling the rib cage up and the diaphragm down. This physical action dramatically increases the volume of the thoracic cavity. According to Boyle's Law, an increase in volume causes an inverse decrease in pressure. This creates a negative pressure gradient inside the lungs compared to the outside atmosphere, effectively vacuuming air down its pressure gradient into the lungs. Exhalation is the passive recoil of these same structures.
Things to Notice about Function and Process
Notice that the How part (the process) requires vastly more details and involves a strict ‘pathway’ approach. It is much more like sequential storytelling compared to the broader, less detailed functional aspects. The more arduous and demanding component of physiology is mastering these detailed processes. This is precisely the reason we need to take our time and fully, deeply understand the fundamentals before we delve into intricate, microscopic details.
What most students quickly recognize about physiology is that it is vastly more conceptual than anatomy because there is almost always a chemical or physical process to describe in a logical, step-by-step manner. There are usually two sides to the functions discussed in physiology. This is because at the very center of the human body is balance, which provides the equilibrium necessary to function properly. When we explain the mechanism of how we breathe in, we must simultaneously understand how we breathe out. When we explain how insulin lowers blood sugar, we must understand how glucagon raises it. Often, once you master one side of the physiological story, the other side falls into place much more easily.
4. Basic Functions of a Complex Organism
Holistically, we will examine Human Physiology as it relates to the foundational basics of how a multi-system, trillions-of-cells living organism functions as a single, perfectly coordinated entity. To be considered a living organism, the body must perform several critical basic functions. Below is an excessively detailed breakdown of these functions:
- Differentiation: The process by which unspecialized, generic cells (like stem cells) develop into highly specialized cells with distinct structures and functions. Extra Example: A single fertilized egg cell divides and its progeny differentiate into vastly different structures, such as conductive nerve cells, contractile muscle cells, and absorptive intestinal cells.
- Responsiveness (Excitability): The body's ability to detect and respond instantly to changes in its internal or external environment. Extra Example: If you touch a hot stove (external stimulus), sensory nerve endings detect the heat and trigger a withdrawal reflex. Internally, if blood calcium levels drop, the parathyroid glands detect this and release hormones to restore balance.
- Metabolism: The sum of all the intricate chemical processes that occur in the body. It consists of two phases: Catabolism (the breaking down of complex molecules into simpler ones to release energy, like digesting food) and Anabolism (the building up of complex molecules from simpler ones, requiring energy, like building new muscle protein).
- Growth and Repair: Growth refers to an increase in body size, either due to an increase in the size of existing cells (hypertrophy), an increase in the total number of cells (hyperplasia), or an increase in the amount of material surrounding cells. Repair is the ongoing process of replacing dead, damaged, or scraped-off cells (like skin cells constantly replacing themselves).
- Movement: Not just walking or running, but motion at every single level. This includes the coordinated action of the entire body, the movement of individual organs (like the stomach churning food or the gallbladder squeezing bile), single cells (like white blood cells physically crawling toward an infection), and even tiny organelles moving inside a cell.
- Excretion: The vital process of removing the toxic by-products of digestion and metabolism. If wastes like urea, carbon dioxide, or lactic acid are allowed to accumulate, they rapidly become fatal. The respiratory, digestive, and urinary systems all share this massive burden.
- Reproduction: The formation of new cells for tissue growth, repair, or replacement, OR the production of a completely new individual to ensure the continuation of the human species.
What we will find is that all of the diverse systems we will study in this course will contain many, if not all, of these basic functions deeply embedded within them.
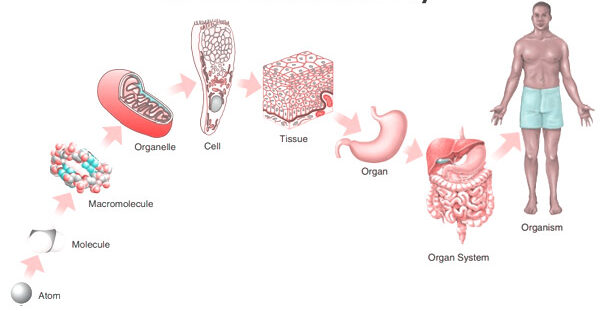
5. Levels of Organization & Body Systems
A body system (also called an organ system) is a highly integrated, complex collection of organs in the body that work seamlessly together to perform a specific, vital function. The truth is that all systems are intimately connected, relying heavily on one another, but it is highly useful for educational purposes to study them separately, even though they are not separate at all. With all of our body systems operating constantly and simultaneously, it is absolutely necessary to have an overarching regulatory system in place to maintain stability, harmony, and equilibrium across all these integrated systems. This unifying, central element in all of physiology is called Homeostasis.
The Six Levels of Structural Organization:
- Chemical Level: The most basic level. Includes atoms (like Carbon, Hydrogen, Oxygen, Nitrogen) combining to form essential molecules (like DNA, glucose, water).
- Cellular Level: Molecules combine to form cells, the basic structural and functional units of an organism. (e.g., Muscle cells, nerve cells, blood cells).
- Tissue Level: Groups of similar cells and their surrounding materials working together to perform a specific function. (The four basic types: Epithelial, Connective, Muscular, Nervous).
- Organ Level: Structures composed of two or more different types of tissues. They have specific functions and recognizable shapes. (e.g., The stomach, heart, liver, brain).
- System Level: Consists of related organs with a common function. (e.g., The digestive system includes the mouth, esophagus, stomach, intestines, liver, and pancreas working to extract nutrients).
- Organismal Level: The highest level. All the parts of the human body functioning together to constitute the total living organism.
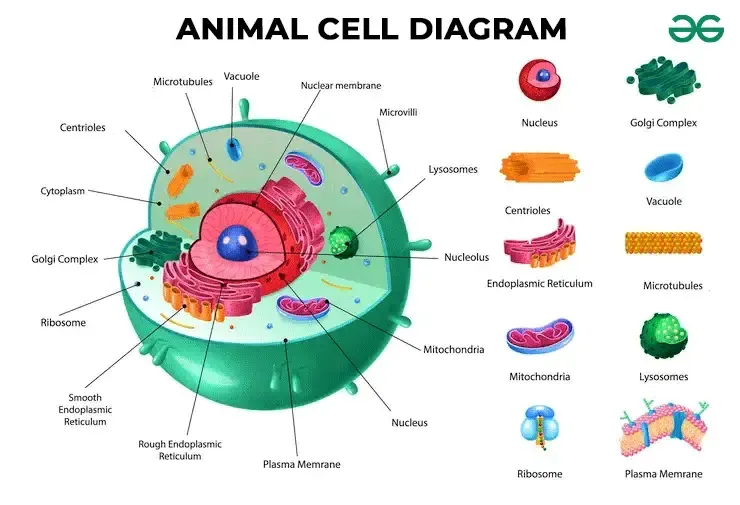
6. The Cell: The Fundamental Unit of Life
The cell is the absolute basic building block of all living things. Every single physiological process begins at the cellular level. To understand how the cell works, we can beautifully compare it to a bustling, highly organized city.
A. The Outer Boundary: The City Wall and Gates
The Cell (Plasma) Membrane
This is the outermost boundary of the cell, an incredibly thin, highly flexible, and selectively permeable (or semipermeable) barrier. It's primarily composed of a phospholipid bilayer, thickly populated with embedded proteins, carbohydrates, and cholesterol molecules.
- Phospholipid Bilayer: Two opposing layers of phospholipids. Each lipid molecule has a hydrophilic ("water-loving") polar head that faces the watery environments inside and outside the cell, and two hydrophobic ("water-fearing") non-polar fatty acid tails that face inward toward each other, forming the water-repelling core of the membrane.
- Proteins: These are crucial for nearly all membrane functions. Integral (transmembrane) proteins span the entire width of the membrane, forming open channels, carrier pumps, and signaling receptors. Peripheral proteins are loosely attached to the inner or outer surface, often involved in intracellular signaling cascades or anchoring the cytoskeleton.
- Cholesterol: Wedged deeply within the hydrophobic core between the fatty acid tails, cholesterol helps stabilize the membrane's fluidity. It prevents the membrane from freezing solid at low temperatures and stops it from becoming too loose and falling apart at high body temperatures.
- Glycocalyx (Carbohydrates): Chains of complex carbohydrates attached to proteins (forming glycoproteins) or attached to lipids (forming glycolipids) on the outer surface. This forms a unique, fuzzy "sugar coat" or cellular "ID tag." Extra Example: The ABO blood group markers on your red blood cells are actually part of the glycocalyx!
Physiological Functions of the Cell Membrane:
- Selective Permeability: Strictly controls exactly what enters and leaves the cell, maintaining perfect homeostasis. (Functions like the city's border control).
- Cell Recognition: The glycocalyx allows immune cells to recognize each other and identify foreign invaders.
- Communication/Signaling: Receptor proteins bind to chemical messengers like hormones and neurotransmitters, translating outside signals into internal actions.
- Cell Adhesion: Proteins allow cells to stick tightly together to form robust tissues (like skin).
- Protection: Provides a flexible physical barrier shielding internal components.
Mnemonic: "People Call Me Protector" for Phospholipids, Cholesterol, Membrane Proteins.
B. The Cell's Internal Environment: The City Hall and Workers
The Cytoplasm
The cytoplasm is literally everything inside the cell membrane but entirely outside the nucleus. It consists of three main elements:
- Cytosol: The jelly-like, semi-fluid, viscous portion where all organelles are suspended. It's mostly water heavily packed with dissolved solutes (ions, glucose, amino acids, ATP, fatty acids, etc.).
- Organelles: The highly specialized "little organs" with very specific, indispensable functions (discussed next).
- Inclusions: Temporary, non-functioning storage bodies. Extra Examples: massive glycogen granules in liver and muscle cells for energy storage, giant lipid droplets in fat cells (adipocytes), and melanin pigment granules in skin cells.
Physiological Functions of the Cytoplasm:
- Site of Many Metabolic Reactions: Crucial biochemical pathways, such as glycolysis (the first, anaerobic step of glucose breakdown to extract energy), occur entirely free-floating in the cytosol.
- Suspension of Organelles: Provides the perfect physical and chemical medium for all organelles to exist, interact, and function.
C. The Control Center: The City Hall/Mayor's Office
The Nucleus
Usually the largest and most prominent organelle, the nucleus is securely enclosed by a double-layered membrane called the nuclear envelope, which is punctured by nuclear pores that highly regulate the entry and exit of molecules. Inside, it contains:
- Chromatin: The relaxed, uncondensed, thread-like form of DNA (our genetic material) carefully wrapped around specialized structural proteins called histones. When the cell prepares to divide, this messy chromatin tightly condenses into distinct, visible structures called chromosomes.
- Nucleolus: A dense, dark, spherical body deeply embedded within the nucleus. It is the primary site of rapid ribosome synthesis.
Physiological Functions of the Nucleus:
- Genetic Control: Contains the cell's entire genetic blueprint (DNA), directing absolutely all cell activities by strictly controlling which proteins are synthesized. (The "master architectural plan" for the city).
- DNA Replication & Transcription: This is where DNA faithfully copies itself before cell division (mitosis) and where DNA's genetic code is safely transcribed into messenger RNA (mRNA), which then leaves the nucleus to give instructions to the factories.
- Ribosome Production: The nucleolus continuously synthesizes and assembles ribosomal RNA (rRNA) subunits.
D. Protein Synthesis and Processing: The Factories and Delivery Services
Ribosomes
Tiny, granular, non-membranous organelles made of ribosomal RNA (rRNA) and various proteins. They are the ultimate "protein factories" of the cell. They physically read the mRNA code coming from the nucleus to perfectly assemble amino acids into long proteins (a process called translation). They can exist as free ribosomes floating in the cytosol (making proteins for immediate use within the cell) or bound ribosomes securely attached to the Endoplasmic Reticulum (making proteins destined for export out of the cell or for embedding into membranes).
Mnemonic: "Ribosomes Read RNA to make Really good pRotein."
Endoplasmic Reticulum (ER)
An extensive, labyrinth-like network of interconnected membranous tubes and flattened sacs that extends massively throughout the cytoplasm, and is directly continuous with the outer membrane of the nuclear envelope.
- Rough Endoplasmic Reticulum (RER): Heavily studded with ribosomes, giving it a "rough" or sandpaper-like appearance under a microscope. Its primary function is to synthesize, fold, and modify proteins that are destined for secretion, insertion into cell membranes, or delivery to lysosomes. (e.g., adding sugar chains in a process called glycosylation). Extra Example: Pancreatic cells that produce massive amounts of digestive enzymes have incredibly large, active RERs.
- Smooth Endoplasmic Reticulum (SER): Completely lacks ribosomes. Its highly specialized functions include massive lipid and steroid hormone synthesis, intense detoxification of harmful drugs and poisons (making it extremely abundant in liver cells), and the storage and release of calcium ions. Extra Example: In skeletal muscle cells, a specialized SER called the sarcoplasmic reticulum stores the calcium absolutely crucial for triggering muscle contraction.
Mnemonic: "Rough ER is Rough on Ribosomes & Really helps Really good pRotein; Smooth ER is Smoothly Synthesizing Steroids & Storing Salcium (calcium) and Speedily Solving Substance Spoilage (detox)."
Golgi Apparatus (Golgi Complex)
A distinct stack of flattened, slightly curved membranous sacs called cisternae. It acts as the ultimate "Post Office" or "Packaging and Shipping Center" of the cell.
Physiological Functions of the Golgi Apparatus:
- Modification, Sorting, and Packaging: It receives vesicles containing raw proteins and lipids from the ER at its cis face, further modifies them (like adding or trimming sugar tags), sorts them based on their final destination, and packages them into new vesicles leaving from its trans face.
- Vesicle Formation: It actively forms various types of vesicles, including secretory vesicles (for exocytosis/dumping out of the cell), lysosomes (cellular stomachs), and transport vesicles that seamlessly deliver new structural components to patch the plasma membrane.
Mnemonic: "Golgi Gathers, Grades, and Gets rid of Garbage (or packages good stuff!)."
E. Energy Production: The Power Plant

Mitochondria
Oval or bean-shaped organelles distinctly enclosed by a double membrane: a relatively smooth outer membrane and an inner membrane that is highly folded inward to form structures called cristae. These folds drastically increase the surface area available for chemical reactions. The dense, fluid-filled space within the inner membrane is called the matrix. Mitochondria also astonishingly possess their own unique, circular DNA (mtDNA) inherited exclusively from the mother.
Physiological Function (The "Powerhouses of the Cell"):
They are the primary site of aerobic cellular respiration. They utilize oxygen to meticulously break down and convert fuel molecules like glucose and fatty acids into ATP (adenosine triphosphate), the primary, usable energy currency of the entire cell. Extra Example: Cells that require massive, continuous amounts of energy, such as cardiac (heart) muscle cells and swimming sperm cells, have spectacularly high numbers of mitochondria compared to inactive cells.
Mnemonic: "Mighty Mitochondria Make Much More Money (ATP)."
F. Waste Management and Recycling: The Cleaning Crew
Lysosomes
Tiny, spherical membranous sacs packed tightly with incredibly powerful hydrolytic (digestive) enzymes that function best in an acidic environment. They act as the "Recycling Centers" and "Stomachs" of the cell.
- They aggressively break down ingested foreign substances and bacteria (phagocytosis).
- They digest worn-out, broken cellular organelles to recycle their raw materials (a process called autophagy).
- In dying or damaged cells, they can burst open to digest the entire cell (autolysis).
- Clinical Correlate: Tay-Sachs disease is a fatal genetic disorder where a specific lysosomal enzyme is missing. Toxic lipids build up uncontrollably in the brain because the lysosomes cannot break them down.
Mnemonic: "Lyso-some = "Lysol" – they lyse (break down) stuff."
Peroxisomes
Smaller membranous sacs containing potent oxidative enzymes, most notably catalase and oxidase. They act as the highly specialized "Detoxification Squad".
- They aggressively neutralize highly reactive, harmful free radicals and detoxify toxins like alcohol (making them very abundant in kidney and liver cells).
- They heavily assist in the breakdown (beta-oxidation) of very long-chain fatty acids for use in energy production.
- During their reactions, they produce dangerous hydrogen peroxide (H₂O₂), but their built-in catalase enzyme instantly converts it safely into water and oxygen.
Mnemonic: "Peroxisomes Produce Peroxide to Purify."
G. The Cell's Internal Support and Movement: The Infrastructure
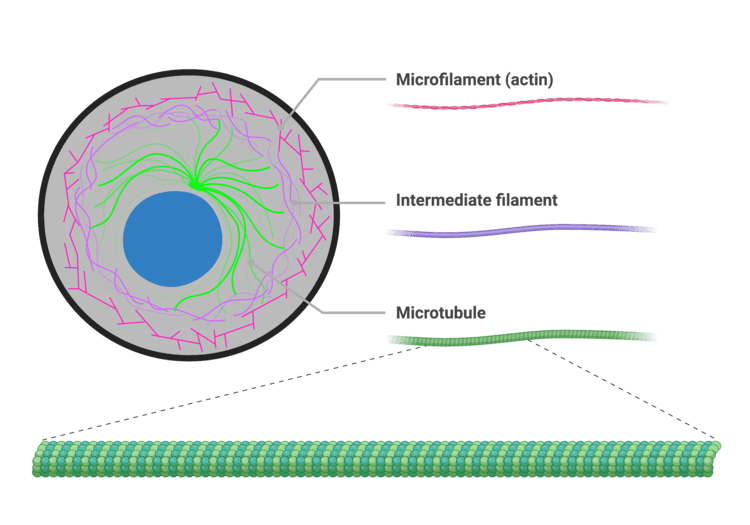
The Cytoskeleton
An intricate, dynamic network of protein filaments extending massively throughout the cytoplasm, providing cell shape, internal support, and physical highways for transport. It consists of three main types of filaments:
- Microfilaments (Actin): The thinnest fibers; deeply involved in cell movement, gross shape changes, cell division (cleavage furrow), and forming the contractile machinery of muscle cells.
- Intermediate Filaments: Tough, rope-like proteins that provide immense structural stability and fiercely resist mechanical pulling stress on the cell. Extra Example: Keratin in skin cells is an intermediate filament that makes skin tough and waterproof.
- Microtubules: The largest elements; hollow tubes made of tubulin. They determine the overall cell shape, form the "railroad tracks" for motor proteins to drag organelles across the cell, and are the primary structural core of cilia, flagella, and the mitotic spindle.
Mnemonic: "Cytoskeleton Supports the Cell Shape and Ships things Swiftly."
Centrosomes, Cilia, and Flagella
- Centrosomes and Centrioles: Located strategically near the nucleus, the centrosome contains two barrel-shaped centrioles positioned at right angles. It acts as the main Microtubule-Organizing Center (MTOC), generating and organizing the mitotic spindle required to pull chromosomes apart during cell division.
- Cilia and Flagella: Hair-like, motile cellular projections entirely made of microtubules.
- Cilia are short and numerous, beating in coordinated waves to move substances across the cell surface. Extra Example: Millions of cilia in your respiratory tract constantly sweep mucus and trapped dust up and out of your lungs.
- Flagella are much longer and usually singular, acting like a whip to propel the entire cell forward. Extra Example: The only human cell with a flagellum is the male sperm cell.
Mnemonic: "Centrosomes and Centrioles Control Cell Civision Carefully."
Summary Table of Organelles
| Organelle |
Key Physiological Functions |
| Plasma Membrane |
Selective barrier, cell recognition, intercellular communication, structural protection. |
| Nucleus |
Ultimate genetic control, houses DNA, site of DNA replication and mRNA transcription. |
| Ribosomes |
Direct protein synthesis via the translation of mRNA into amino acid chains. |
| Rough ER (RER) |
Synthesis, folding, and intense modification of proteins destined for export or membrane insertion. |
| Smooth ER (SER) |
Lipid and steroid synthesis, detoxification of drugs/poisons, massive intracellular Ca²⁺ storage. |
| Golgi Apparatus |
Modifies, sorts, meticulously packages proteins and lipids into transport/secretory vesicles. |
| Mitochondria |
Site of aerobic cellular respiration, massive ATP synthesis (the ultimate powerhouse). |
| Lysosomes |
Intracellular digestion of debris, bacteria, and old organelles; primary cellular waste removal. |
| Peroxisomes |
Detoxification (especially neutralizing free radicals and alcohol), extensive fatty acid breakdown. |
| Cytoskeleton |
Maintains cell shape, structural support, internal transport tracks, enables cell motility. |
| Centrosomes |
Organize the microtubule network and generate the mitotic spindle during cell division. |
| Cilia / Flagella |
Cilia move external substances across the cell surface; Flagella physically propel the entire cell. |
7. Biological Membranes: The Fluid Mosaic Model
Biological membranes are highly dynamic, incredibly fluid structures that sharply define the boundaries of cells (plasma membrane) as well as the internal organelles. They are absolutely essential for maintaining cellular integrity, strictly regulating transport, facilitating communication, and housing vital enzymatic reactions. The most universally accepted scientific model describing membrane structure is the Fluid Mosaic Model.
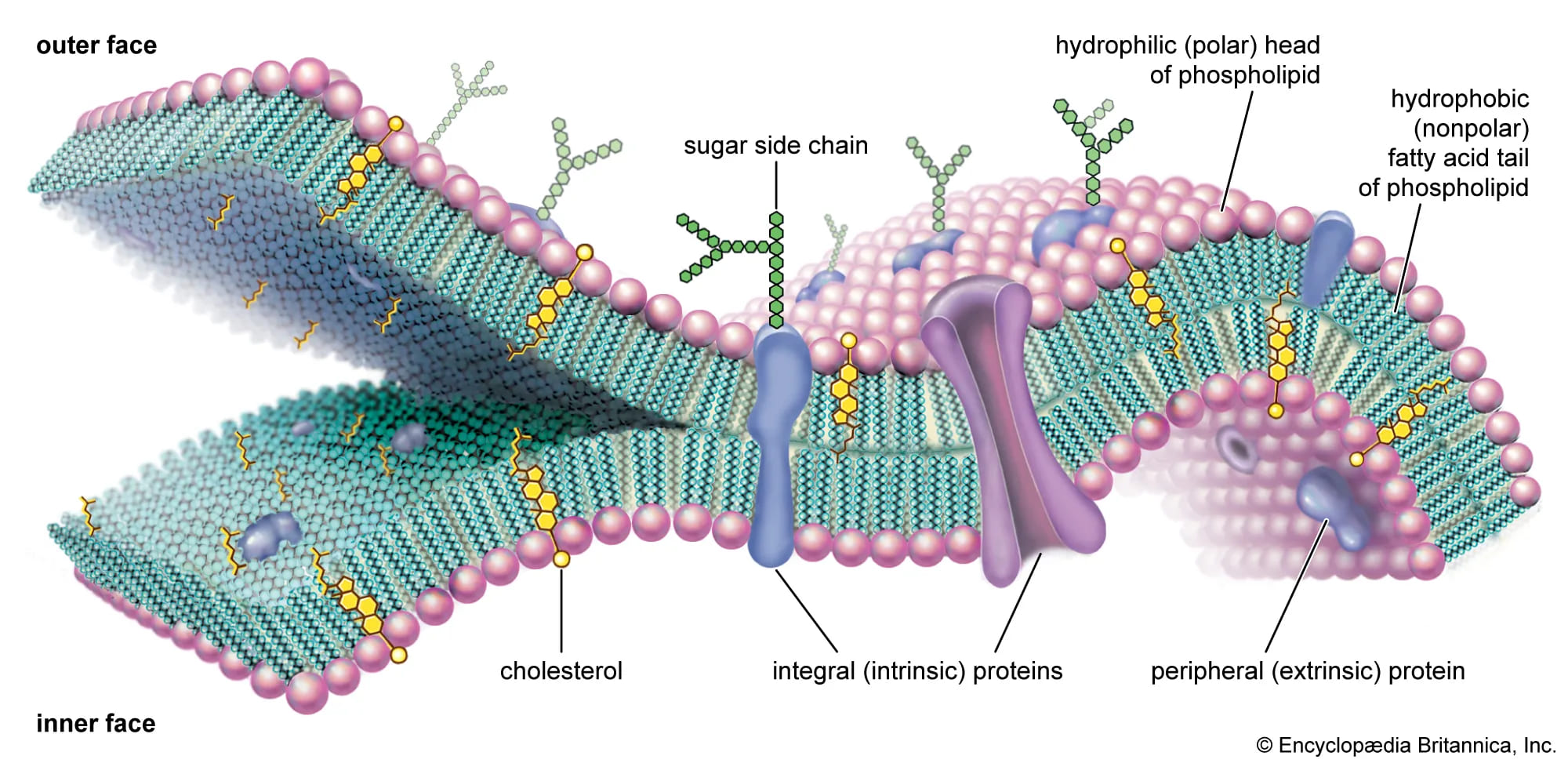
The Fluid Mosaic Model
Proposed brilliantly by Singer and Nicolson in 1972, this model describes the cell membrane as a highly fluid lipid bilayer where an array of proteins are embedded or attached, looking much like a complex, ever-shifting mosaic artwork.
- "Fluid": Refers to the constant, rapid movement of individual phospholipid molecules and proteins within the two-dimensional plane of the membrane. Lipids and many proteins are not locked in place; they constantly drift laterally, rotate on their axes, and flex their tails.
- "Mosaic": Refers to the diverse, scattered "patchwork" of different functional proteins and other structural molecules (like cholesterol and carbohydrates) permanently or temporarily embedded within the vast ocean of the lipid bilayer.
A. Lipids of the Cell Membrane
The central, primary structural framework of the membrane is the fluid lipid bilayer, which is predominantly made of phospholipids and cholesterol.
1. Phospholipids
Phospholipids are by far the most abundant lipids in the membrane. They possess a crucial chemical property: they are amphipathic. This means a single molecule contains both a highly hydrophilic (water-loving) polar head and two highly hydrophobic (water-fearing) non-polar fatty acid tails. In the watery environment of the body, they spontaneously and instantly self-assemble to form a bilayer. The hydrophobic tails hide inward, desperately avoiding water, while the hydrophilic heads proudly face the watery environments inside (intracellular fluid) and outside (extracellular fluid) the cell.
2. Cholesterol
Cholesterol molecules are tough, rigid, ring-shaped lipids inserted tightly between the phospholipids. They act as the ultimate membrane temperature buffer, perfectly regulating fluidity. At normal, warm body temperatures, cholesterol restricts movement, reducing excessive fluidity and making the membrane stronger and less leaky. Conversely, at dangerously low temperatures, cholesterol prevents the phospholipid tails from packing too tightly together, thus increasing fluidity and stopping the cell membrane from freezing solid.
Lipid Functions in the Cell Membrane:
- Forms the fundamental, self-healing bilayer structure.
- Provides a robust selectively permeable barrier, naturally allowing only small, uncharged, fat-soluble substances (O₂, CO₂, steroid hormones) to easily pass through directly.
- Acts as an impenetrable physical barrier for all water-soluble substances (glucose, salts, ions), which absolutely require physical assistance from embedded proteins to cross.
B. Membrane Proteins
While lipids form the barrier, proteins are the hardworking machines of the membrane, performing almost all of its specific, dynamic functions.
1. Integral (Transmembrane) Proteins
These are tightly bound proteins that fully span across the entire thickness of the membrane. They are deeply embedded in the hydrophobic core and can only be removed by completely destroying the bilayer. They primarily function as open channels, carrier transport proteins, active pumps, receptors, and enzymes.
2. Peripheral Proteins
These are loosely, temporarily bound to the membrane's inner or outer surface. They do not penetrate the hydrophobic core and are easily detached without harming the membrane. They often function as localized enzymes, signaling relays, or cytoskeletal anchors holding the membrane shape.
Functions of Membrane Proteins:
- Transport: Facilitating the precise movement of specific, rejected substances across the barrier (channels, carriers, ATP pumps).
- Enzymatic Activity: Catalyzing vital metabolic reactions directly at the membrane surface.
- Signal Transduction: Acting as sophisticated receptors catching chemical messengers (like adrenaline) and transmitting the order inside.
- Cell-Cell Recognition: Acting as strict identification tags (glycoproteins) so immune cells don't attack the body's own tissues.
- Intercellular Joining: Forming tight junctions and desmosomes to physically link neighboring cells together.
- Attachment to Cytoskeleton & ECM: Providing immense structural stability by tethering the cell's skeleton to the outside world.
C. Carbohydrates of the Cell Membrane
Carbohydrates are exclusively found on the external, outward-facing surface of the plasma membrane. They never face the inside. They are permanently attached to lipids (forming glycolipids) or to proteins (forming glycoproteins). This entire "sugar coat" covering the cell is called the glycocalyx, which serves as a highly specific, unique molecular signature for every different cell type in your body.
Functions of Membrane Carbohydrates (Glycocalyx):
- Cell-Cell Recognition: Crucial for distinguishing "self" tissues from "non-self" invaders (e.g., orchestrating immune responses, dictating ABO blood types, and organ transplant rejection).
- Cell Adhesion: The sticky sugars help cells firmly bind to one another in tissues.
- Receptors: Can uniquely act as receptors for specific hormones, or tragically, as hijacking points for bacterial toxins and viruses (like the flu or COVID-19).
- Protection: Provides a cushioning, protective physical barrier against mechanical damage and harsh enzymes.
Properties of the Cell Membrane
The specific composition and arrangement of lipids, proteins, and carbohydrates endow the cell membrane with its most essential, life-sustaining properties:
- Selectively Permeable (Semi-permeable): This is arguably its most important property. The membrane precisely and selfishly regulates exactly which substances can enter or leave the cell. The hydrophobic lipid core acts as the primary absolute barrier. Small, nonpolar molecules (O₂, CO₂) and highly lipid-soluble molecules pass directly and effortlessly. However, charged ions (Na⁺, K⁺) and large polar molecules (glucose) are utterly rejected and require specific, designated transport proteins to grant them entry.
- Fluidity: The membrane is absolutely not a rigid, solid shell; its molecular components are in constant, swirling motion. Fluidity is heavily influenced by body temperature, the amount of cholesterol (which acts as a stabilizing buffer), and the degree of saturation of the fatty acid tails (kinked, unsaturated tails increase fluidity). This property is absolutely essential for vesicles to fuse with the membrane, for the cell to divide, and for transport proteins to physically shift their shapes.
- Asymmetry: The two faces (the inner leaflet touching the cytoplasm, and the outer leaflet touching the blood/fluid) of the plasma membrane are completely structurally and functionally different. For example, carbohydrates are exclusively on the outer surface (glycocalyx), and specific lipids and signaling proteins are oriented strictly in one direction. This is vital for directional signaling, shape maintenance, and cell recognition.
- Self-Sealing Capability: Due to the powerful thermodynamic forces of hydrophobic interactions, if the membrane is punctured or torn, the lipids have a natural, spontaneous tendency to instantly re-seal themselves, completely preventing the fatal leakage of cytoplasmic contents. This is crucial for maintaining cell integrity during physical stress or when a needle enters a cell.
Summary of Key Membrane Functions:
- Protective Barrier: Encloses the cell's delicate contents, safely separating the intracellular from the chaotic extracellular environment.
- Selective Transport: Heavily regulates the passage of every single substance entering and exiting the cell.
- Cell-Cell Communication: Contains specific receptors for circulating hormones, drugs, and neurotransmitters.
- Cell Recognition & Adhesion: Facilitates cell identification by the immune system and the formation of solid tissues.
- Enzymatic Activity: Houses membrane-bound enzymes that rapidly catalyze specific biochemical reactions.
- Maintenance of Cell Shape: Provides profound structural support in conjunction with the inner cytoskeleton.
- Generates Membrane Potential: Crucial for the electrical firing of nerve and muscle cells.
- Endocytosis & Exocytosis: Manages the bulk, massive transport of large materials into and out of the cell.
8. Membrane Potential: The Electrical Voltage Across the Membrane
Before looking at exactly how things physically move across the membrane, it's absolutely essential to understand that there is a permanent electrical difference, or voltage, sitting directly across every single cell membrane in your body. This is called the membrane potential.
Membrane Potential is the exact measurable difference in electrical charge (or potential energy) between the inside and the outside of a cell. By strict scientific convention, the inside of the cell is always measured as being negative relative to the outside.
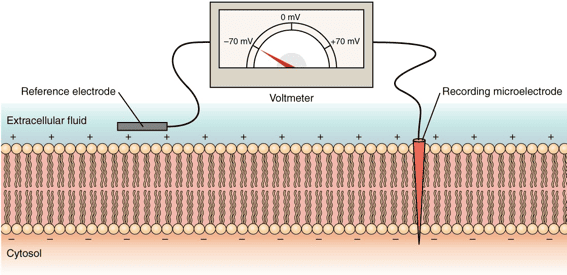
How is the Membrane Potential Established?
It is driven by three distinct, constantly working factors:
- Unequal Distribution of Ions: There are radically different concentrations of ions (charged particles) inside versus outside the cell.
- Outside the cell (Extracellular Fluid - ECF): Features a massively high concentration of Na⁺ (sodium) and Cl⁻ (chloride).
- Inside the cell (Intracellular Fluid - ICF): Features a massively high concentration of K⁺ (potassium) and huge, negatively charged proteins and phosphates (which are physically too large to ever leave the cell, trapping their negative charge inside).
- Selective Permeability of the Membrane: The cell membrane is not equally permeable to all ions. At rest, it is packed with specific "leak channels" that make it vastly more permeable to K⁺ than to Na⁺. Because K⁺ is highly concentrated inside, it constantly leaks out down its concentration gradient. Every time a positive K⁺ leaves, it leaves behind a negative charge, making the inside of the cell significantly more negative.
- Sodium-Potassium Pump (Na⁺/K⁺ ATPase): This relentless active transport pump burns massive amounts of ATP to constantly eject exactly 3 Na⁺ ions out of the cell for every 2 K⁺ ions it pumps back in. Because it pumps out more positive charges (3) than it brings in (2), this pump is inherently electrogenic and contributes directly and continuously to the negative charge inside the cell.
Resting Membrane Potential
In a resting (unstimulated) neuron or muscle cell, the steady-state electrical potential established and maintained perfectly by these factors is called the Resting Membrane Potential. It is typically measured at around -70 mV (millivolts), meaning the inside is 70 millivolts more negative than the outside.
Physiological Significance:
The resting membrane potential is absolutely not just a passive, resting state; it is a massive form of stored, coiled potential energy (like a stretched rubber band) crucial for:
- Excitability: It allows excitable cells (like neurons and heart muscle cells) to instantly open gates and generate rapid, explosive electrical signals (action potentials) for instant communication, thought, and muscle contraction.
- Secondary Active Transport: The immense energy stored in the steep Na⁺ and K⁺ ion gradients can be brilliantly harnessed by the cell to power the "uphill" transport of other essential substances (like glucose and amino acids) across the membrane.
Clinical Correlate: Hyperkalemia
If a patient's kidneys fail, potassium (K⁺) builds up in the blood outside the cell (a condition called hyperkalemia). This destroys the carefully maintained K⁺ gradient. The resting membrane potential is ruined, the heart muscle cells cannot reset their electrical charge, and the patient suffers a sudden, fatal cardiac arrhythmia (heart attack).
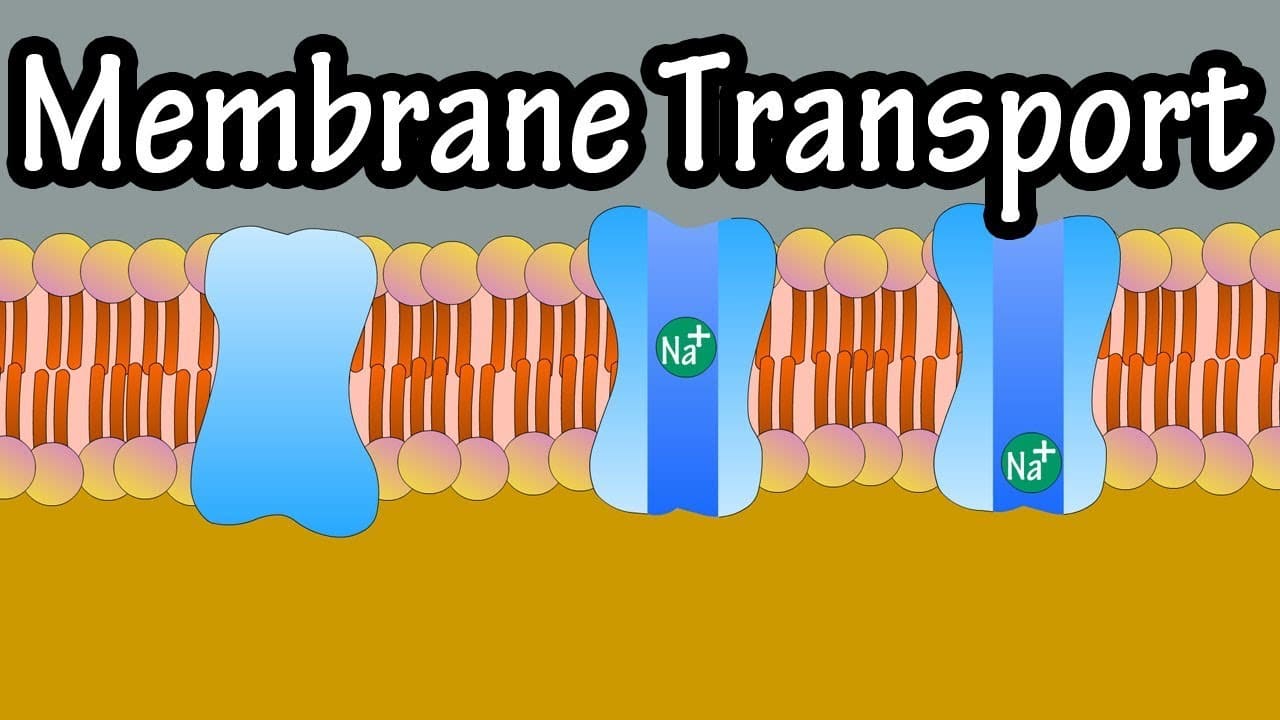
9. Membrane Transport
Membrane transport is the fundamental physiological process that meticulously governs the movement of all substances across biological membranes. It's essential for maintaining cellular homeostasis, acquiring vital nutrients, aggressively expelling toxic waste products, and facilitating cell-to-cell communication. Substances cross the membrane via two general, overarching mechanisms: Passive Transport and Active Transport.
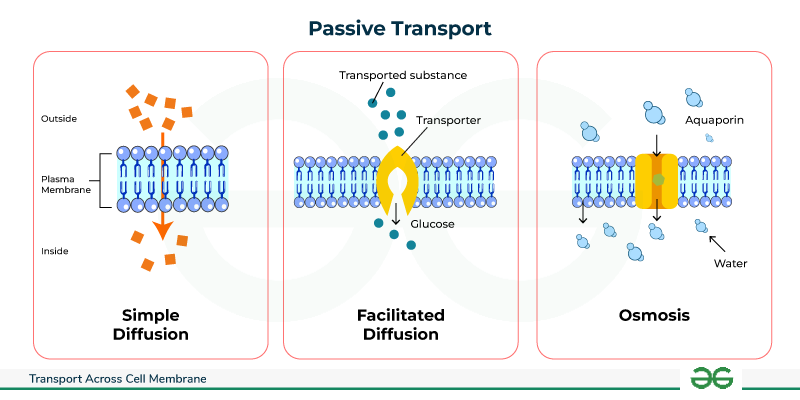
1. Passive Transport: Moving Downhill
Passive transport is the movement of substances across a cell membrane completely without the direct expenditure of cellular metabolic energy (ATP). This movement is always "downhill," meaning down the electrochemical or concentration gradient of the substance. The energy driving this movement comes purely from the inherent, random kinetic energy of the molecules themselves and the potential energy stored in the concentration gradient.
1.1. Simple Diffusion: Through the Lipid Bilayer
In simple diffusion, substances move directly and effortlessly through the lipid bilayer without any help from membrane proteins whatsoever.
- Highly Permeable: Small, nonpolar (lipophilic/fat-soluble) molecules like O₂, CO₂, and steroid hormones readily dissolve directly into the hydrophobic core and pass completely through to the other side.
- Moderately Permeable: Small, uncharged polar molecules like water and ethanol can pass to a very limited degree because they are small enough to slip through the lipid gaps.
- Impermeable: Large polar molecules (like glucose) and absolutely all charged ions (Na⁺, K⁺, Ca²⁺) cannot pass through the lipid core on their own under any circumstances.
The sole driving force is the concentration gradient. The random, chaotic molecular motion (kinetic energy) results in a net movement from an area of highly crowded concentration to an area of lower concentration, continuing unabated until perfect equilibrium is reached across the membrane.
Key Characteristics of Simple Diffusion:
- Absolutely no membrane proteins are involved.
- Does not exhibit saturation kinetics: The rate of diffusion increases linearly forever with the concentration gradient; there is no maximum transport rate (Vmax) because there are no proteins to get "full".
- According to Fick's Law, the rate is directly proportional to the gradient magnitude, lipid solubility, and membrane surface area, and inversely proportional to molecular size and the physical thickness of the membrane.
1.2. Facilitated Diffusion: Protein-Assisted Passage
This process uses specialized integral membrane proteins (channels or carriers) to beautifully facilitate the movement of specific, impermeable substances down their electrochemical gradient. It is still completely passive, as absolutely no ATP is directly consumed.
A. Channel Proteins (Pores)
These proteins form a hollow, water-filled pore directly across the membrane, allowing the incredibly rapid, single-file passage of specific ions or water molecules. Most channels are tightly gated, meaning they act like doors that only open or close in response to specific, required stimuli:
- Voltage-Gated Channels: Respond instantly to changes in electrical membrane potential (e.g., Na⁺ and K⁺ channels crucial in firing neurons).
- Ligand-Gated Channels: Respond only when a specific chemical messenger binds to them (e.g., neurotransmitter receptors at muscle synapses).
- Mechanically-Gated Channels: Respond directly to physical deformation or stretching of the membrane (e.g., touch receptors in your skin, or hearing receptors in the ear).
- Leak Channels: Are generally always randomly fluttering open and closed, heavily contributing to the resting membrane potential.
- Extra Examples: Ion channels (Na⁺, K⁺, Cl⁻, Ca²⁺) and aquaporins, which are highly specialized channels dedicated exclusively to letting massive amounts of water through instantly.
B. Carrier Proteins (Transporters)
These proteins work like revolving doors. They physically bind to a specific molecule on one side, undergo a massive conformational (shape) change, and then release the molecule safely on the other side. This physical shifting process makes it much slower than channel-mediated transport.
- Saturation Kinetics: Because there are a strictly finite number of carrier proteins on a cell, the transport rate has an absolute maximum speed limit (Vmax) that hits when every single carrier is occupied and working as fast as it can.
- Specificity: Carriers are incredibly highly specific for the exact molecule(s) they are designed to transport.
- Competition: Structurally similar, "imposter" molecules can compete for the exact same binding site, blocking the real molecule.
- Extra Examples: Glucose Transporters (GLUT proteins, which insulin commands to surface) and various amino acid transporters.
Key Characteristics of Facilitated Diffusion:
- Strictly involves specific integral membrane proteins (channels or carriers).
- Exhibits saturation kinetics (Vmax limit) due to the limited, physical number of transporters.
- Can be easily subject to chemical competition.
- Can be heavily regulated by the cell (e.g., by gating channels shut, or inserting/removing carriers from the membrane to control absorption).
1.3. Osmosis: The Grand Movement of Water
Osmosis is the incredibly powerful net movement of water across a selectively permeable membrane, strictly from an area of higher water concentration (which means lower solute concentration) directly to an area of lower water concentration (which means higher, saltier solute concentration). The driving force is the water potential gradient, determined entirely by the difference in non-penetrating solute concentration on either side of the membrane.
Osmotic pressure is the "pulling" or "sucking" force a solution with a higher solute concentration forcefully exerts to draw water toward it. Tonicity refers to the actual physical effect of a surrounding solution on the cell's total volume:
- Isotonic: The concentration is perfectly equal inside and outside. There is no net water movement; cell volume remains perfectly normal.
- Hypotonic: The outside fluid is dilute (watery). Water rushes rapidly into the cell, causing it to aggressively swell and potentially burst (a fatal event called lysis).
- Hypertonic: The outside fluid is extremely concentrated (salty). Water is sucked rapidly out of the cell, causing the cell to drastically shrivel and shrink (a process called crenation).
Clinical Correlate: Intravenous (IV) Fluids
When a patient arrives at the hospital dehydrated, you cannot simply inject pure, hypotonic tap water into their veins. Doing so would cause their red blood cells to rapidly absorb the water, swell, and violently explode (hemolysis), killing the patient. Instead, nurses administer 0.9% Normal Saline, which is perfectly isotonic to human blood plasma, safely rehydrating the body without destroying the cells.
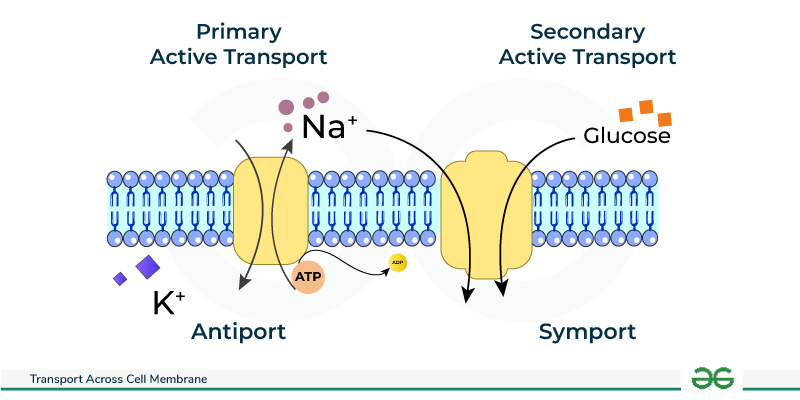
2. Active Transport: Against the Current, with Energy
Active transport is the strenuous, demanding process of actively moving substances across a cell membrane directly against their electrochemical or concentration gradient (i.e., dragging them from a region of lower concentration forcefully into a region of higher concentration). This distinctly "uphill" movement necessitates the direct or indirect expenditure of precious cellular metabolic energy, almost invariably derived from the violent hydrolysis of ATP.
2.1. Primary Active Transport: Direct ATP Expenditure
Primary active transporters are robust integral membrane proteins that function fundamentally as ATPases; they directly bind to and snap the phosphate off of ATP, utilizing that massive burst of released energy to physically power the forced movement of solutes. These heavy-duty transporters are often called "pumps."
- Na⁺/K⁺ ATPase (Sodium-Potassium Pump): Found aggressively pumping in virtually all animal cells, this absolutely vital, life-sustaining pump forcibly moves 3 Na⁺ ions entirely out of the cell and drags 2 K⁺ ions back into the cell for every single ATP molecule hydrolyzed. It is strictly electrogenic (creates a negative charge imbalance) and is undeniably fundamental for maintaining steep Na⁺/K⁺ gradients, establishing the baseline resting membrane potential, regulating water balance and cell volume, and driving secondary active transport.
- Ca²⁺ ATPases (e.g., SERCA, PMCA): These relentless pumps maintain the extremely, dangerously low intracellular Ca²⁺ concentration needed for survival. SERCA constantly pumps Ca²⁺ safely into the sarcoplasmic/endoplasmic reticulum for deep storage (which is absolutely crucial for allowing a muscle to relax after a contraction), while PMCA furiously pumps Ca²⁺ completely out of the cell membrane into the blood.
- H⁺/K⁺ ATPase (Gastric Proton Pump): Located almost exclusively in the specialized parietal cells of the stomach lining, this pump aggressively secretes H⁺ (acid protons) directly into the stomach lumen, creating the highly destructive, acidic environment (pH 1-2) necessary for dissolving food and activating digestion. Extra Example: This exact pump is the sole target of Proton Pump Inhibitor (PPI) drugs, like Omeprazole, which shut the pump down to cure severe stomach ulcers and acid reflux.
- ABC Transporters (ATP-Binding Cassette): A gigantic, ancient superfamily of transporters that move a vast, diverse array of substrates.
Extra Example 1: MDR1 (P-glycoprotein) is an ABC transporter notorious in oncology; it causes multidrug resistance in cancer cells by acting as a biological bouncer, actively catching and pumping expensive chemotherapy drugs right back out of the tumor cell before they can work.
Extra Example 2: The CFTR protein is a specialized Cl⁻ channel; a genetic mutation destroying this exact pump causes the thick, deadly mucus buildup characteristic of the disease Cystic Fibrosis.
2.2. Secondary Active Transport (Co-transport)
Secondary active transport is highly efficient because it does not directly hydrolyze ATP itself. Instead, it ingeniously acts like a waterwheel; it uses the immense potential kinetic energy stored in an existing, steep electrochemical gradient (typically the massive Na⁺ gradient that was previously created and paid for by the Na⁺/K⁺ pump) to power the transport of a completely second substance against its own gradient.
- Symporters (Cotransporters): Both the driving ion (e.g., Na⁺ rushing down its gradient) and the "freeloading" transported solute are physically dragged across the membrane in the exact same direction. Extra Example: The Na⁺-Glucose Symporter (SGLT) found heavily in the intestine and kidneys, which utilizes the rushing flow of Sodium to forcefully absorb essential Glucose from your food against a steep gradient, ensuring none is wasted in the feces or urine.
- Antiporters (Exchangers): The driving ion rushes in, providing energy to forcefully eject the transported solute in the exact opposite direction. Extra Example: The Na⁺-Ca²⁺ Exchanger (NCX), which is absolutely crucial for aggressively removing toxic excess Ca²⁺ from cardiac (heart) muscle cells after every single heartbeat, allowing the heart to relax. Also, the Na⁺-H⁺ Exchanger (NHE) which pumps acid out of the cell to regulate intracellular pH.
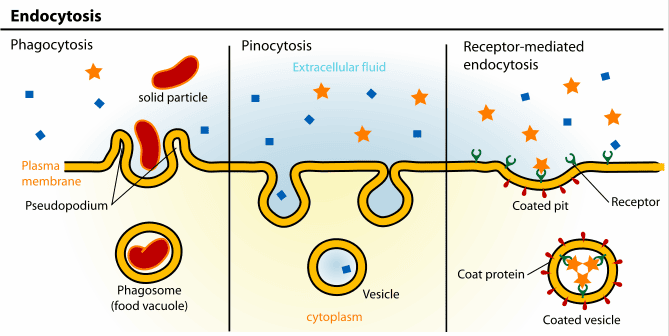
3. Vesicular Transport (Bulk Transport): For the Heavy Lifting
Vesicular transport is the macro-scale mechanism used exclusively for moving massively large molecules, whole macromolecules, and giant particulate matter (even whole bacteria) into or out of the cell. It involves the complex, energy-demanding physical formation, movement, and fusion of large membrane-bound sacs called vesicles. Because it physically reshapes the cell membrane, it absolutely always requires massive amounts of cellular energy (ATP).
3.1. Endocytosis: Bringing the Outside In
Endocytosis is the active process by which cells internalize large substances. The plasma membrane physically sinks inward (invaginates), wraps entirely around the material, and pinches off, forming a brand-new intracellular vesicle.
- Phagocytosis ("Cell Eating"): The aggressive, targeted ingestion of massive, solid particles like invading bacteria, dead tissue, or cellular debris by highly specialized immune sentinels (e.g., macrophages and neutrophils). The cell physically extends long "arms" called pseudopods to totally engulf the target, forming a giant internal death-chamber called a phagosome, which then merges with a lysosome for total destruction.
- Pinocytosis ("Cell Drinking"): The constant, non-specific, routine gulping of tiny droplets of extracellular fluid and whatever dissolved solutes happen to be floating in it. It is how cells sample their environment.
- Receptor-Mediated Endocytosis: An exquisitely, highly specific, targeted process. Specific extracellular ligands (e.g., LDL-cholesterol particles, iron-carrying transferrin, or even sneaky viruses) must perfectly bind to complementary, matching receptors on the surface. These loaded receptors then rapidly slide together and cluster into specialized depressions called clathrin-coated pits, which fold inward and are cleanly brought into the cell as a customized vesicle.
3.2. Exocytosis: Releasing to the Outside
Exocytosis is the reverse process by which cells actively release large substances. Intracellular, membrane-bound vesicles are driven by motor proteins to the edge of the cell, where they seamlessly fuse with the plasma membrane, violently dumping their contents to the outside world.
- Constitutive Secretion: A continuous, ongoing, unregulated baseline process operating in all cells. It is used to constantly deliver newly minted lipids and proteins to expand the plasma membrane, and to continuously secrete the structural components of the extracellular matrix (like collagen).
- Regulated Secretion: A highly controlled process that occurs exclusively in specialized secretory cells (e.g., brain neurons, pancreatic endocrine cells). Secretory vesicles containing highly potent products like neurotransmitters, digestive enzymes, or hormones are stockpiled safely near the membrane. They are strictly held back and only released in a massive burst in response to a very specific, targeted signal (almost always a sudden, sharp rise in intracellular Ca²⁺).
10. The "Why": Importance and Functions of Membrane Transport
The precise, unrelenting control over exactly what enters and exits a cell underlies the success of virtually every single physiological process in the entire human body.
- Maintenance of Cellular Homeostasis: Strict, life-or-death ion gradients (e.g., maintaining low intracellular Na⁺ and high K⁺) and exact internal pH are aggressively maintained at incredibly great energy cost to absolutely prevent cell death and ensure proper, flawless enzyme function.
- Nutrient Acquisition: Transport systems enable cells to highly efficiently scavenge, absorb, and aggressively concentrate essential, life-sustaining molecules like glucose, vitamins, and amino acids from the blood.
- Waste Removal: Active transporters continuously expel harmful, toxic metabolic waste products (like urea, lactic acid, and excess acid protons), totally preventing their lethal accumulation inside the cytoplasm.
- Generation of Electrical Signals: In complex neurons and muscle cells, the rapid, highly controlled movement of ions surging through gated channels generates action potentials, which are the fundamental electrical basis of every single thought, heartbeat, and physical movement.
- Cell-to-Cell Communication: Exocytosis violently releases neurotransmitters across synapses and dumps hormones into the blood, while endocytosis helps regulate receptor sensitivity by swallowing up overused receptors from the membrane.
- Regulation of Cell Volume: Constant ion pumping, most especially the tireless Na⁺/K⁺ ATPase, strictly controls internal intracellular osmolarity, absolutely preventing cells from fatally swelling up and bursting or aggressively shrinking and collapsing.
- Absorption and Reabsorption: Massive, highly coordinated transport processes in the Gastrointestinal (GI) tract and the nephrons of the kidneys are utterly essential for absorbing nutrients from food and perfectly regulating the entire body's water, total electrolyte, and systemic acid-base balance.
Summary of Membrane Transport Mechanisms
| Process |
Energy Requirement |
Gradient Direction |
Transporter Requirement |
What Specifically Moves? |
Examples & Clinical Notes |
| Passive Processes (No ATP Burned) |
| Simple Diffusion |
None |
Downhill (High to Low) |
None (Straight through lipid) |
Small, highly lipid-soluble molecules |
O₂, CO₂, steroid hormones crossing tissues. |
| Facilitated Diffusion |
None |
Downhill (High to Low) |
Yes (Channel or Carrier protein) |
Ions, large glucose, amino acids |
Glucose transporters (GLUT), Na⁺/K⁺ voltage channels. |
| Osmosis |
None |
Downhill (High water to Low water) |
Yes (Aquaporins heavily aid) |
Water molecules exclusively |
Red blood cells expanding or shrinking in different tonic IV solutions. |
| Active Processes (Requires ATP) |
| Primary Active Transport |
Yes (Directly breaks ATP) |
Uphill (Low to High) |
Yes (A specialized Pump) |
Ions against steep gradients |
Na⁺/K⁺ pump, Ca²⁺ SERCA pump, H⁺ stomach acid pump. |
| Secondary Active Transport |
No (Uses kinetic energy of an existing ion gradient) |
Uphill (Low to High) |
Yes (Symporter or Antiporter) |
Ions, glucose, amino acids dragged along |
Na⁺-glucose co-transporter (SGLT) in the kidney. |
| Vesicular Transport |
Yes (Massive ATP used) |
N/A (Bulk physical movement) |
No (Uses membrane vesicles) |
Large particles, whole bacteria, macromolecules, bulk fluids |
Phagocytosis (macrophages eating bacteria), Exocytosis (releasing insulin). |
List of Academic References
- Hall, J. E., & Hall, M. E. (2020). Guyton and Hall Textbook of Medical Physiology (14th ed.). Elsevier. (Definitive source on membrane potentials, the Na⁺/K⁺ pump, and systemic physiology).
- Boron, W. F., & Boulpaep, E. L. (2016). Medical Physiology (3rd ed.). Elsevier. (Excellent deep dive into cellular organelles, secondary active transport, and molecular physiology).
- Alberts, B., Johnson, A., Lewis, J., et al. (2014). Molecular Biology of the Cell (6th ed.). Garland Science. (The absolute gold standard for the Fluid Mosaic Model, vesicular transport, and cytoskeletal structures).
- Costanzo, L. S. (2017). Physiology (6th ed.). Elsevier. (Highly recommended for clear, mechanistic explanations of functional vs. process physiology and simple diffusion mathematics).
- Chabner, L. (2020). The Language of Medicine (12th ed.). Saunders. (A premier resource for medical terminology, etymology, roots, and prefixes like pancytopenia).