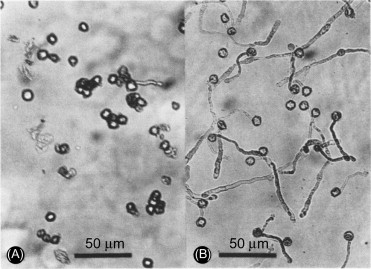
Coccidioides & Paracoccidioides brasiliensis
Coccidioides and Paracoccidioides
By the conclusion of this exhaustive master guide, you will be deeply conversant with:
- The complete microbiological lifecycles of Coccidioides spp. and Paracoccidioides brasiliensis.
- The profound impact of environmental factors, geology, and endocrinology (e.g., estrogen) on the pathogenesis of these fungi.
- The distinct immunological responses (Th1 vs. Th2) required for host defense and granuloma formation.
- The clinical manifestations, diagnostic "buzzwords" (Spherules vs. Pilot Wheels), and pharmacological treatments for these severe systemic mycoses.
Part 1: Coccidioides species (Valley Fever)
I. Introduction to Coccidioides
Coccidioides species are the highly virulent causative agents of the systemic fungal infection universally known as Coccidioidomycosis (commonly referred to clinically as "Valley Fever", "San Joaquin Valley Fever", or "Desert Rheumatism").
- Historical & Clinical Context: It has been recognized in medical literature for over a century but is currently classified as a major reemerging fungal infection.
- Drivers of Reemergence: The resurgence of Coccidioidomycosis can be attributed to several compounding factors:
- Overpopulation, rapid urban development, and construction in highly endemic desert areas (disturbing the soil).
- Compromised cellular immunity across the population (especially exacerbated by the rise of the HIV/AIDS epidemic, organ transplantation, and immunosuppressive biologic drugs).
- Advances in the prevention and treatment of other fungal and bacterial infections (leaving an opportunistic ecological gap).
Bioterrorism Potential & Biosafety Hazard
The emergence of Coccidioides spp. as potential agents of bioterrorism is a major public health concern.
Physiology Expansion: Because the infective dose is extraordinarily low—as few as ONE single spore—and the spores are incredibly durable and easily aerosolized over vast distances, it is federally classified as a highly dangerous Biosafety Level 3 (BSL-3) pathogen. Routine laboratory handling outside of strict biocontainment has historically led to massive, fatal laboratory outbreaks.
II. Mycology & Taxonomic Classification
Classification (By Molecular Analysis):
Kingdom: Fungi ➔ Phylum: Ascomycota ➔ Class: Eurotiomycetes ➔ Order: Onygenales ➔ Family: Onygenaceae ➔ Genus: Coccidioides.
- Species Delineation: There are two identical sibling species: C. immitis (geographically localized mostly to the San Joaquin Valley of California) and C. posadasii (found outside California, such as Arizona, Texas, Mexico, and South America). They are clinically and morphologically indistinguishable.
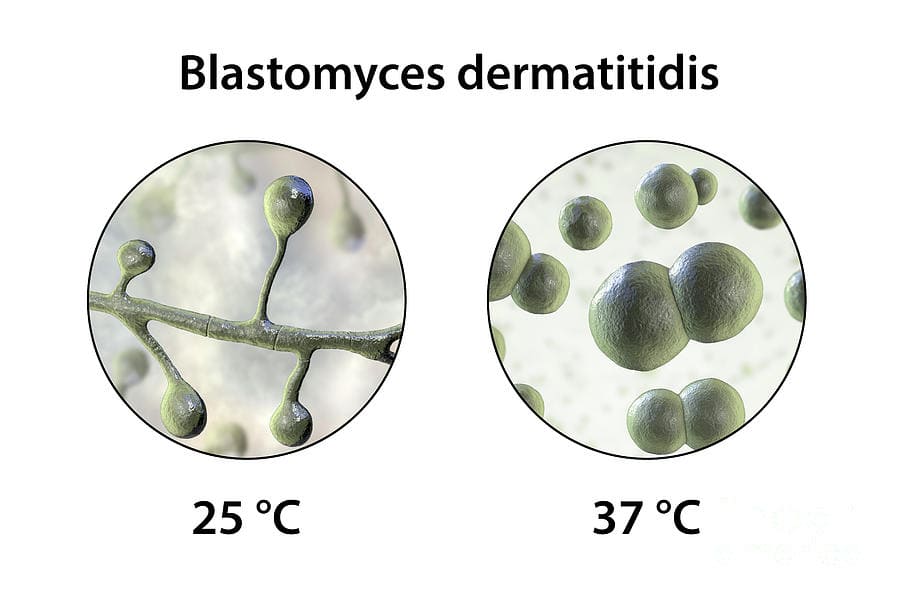
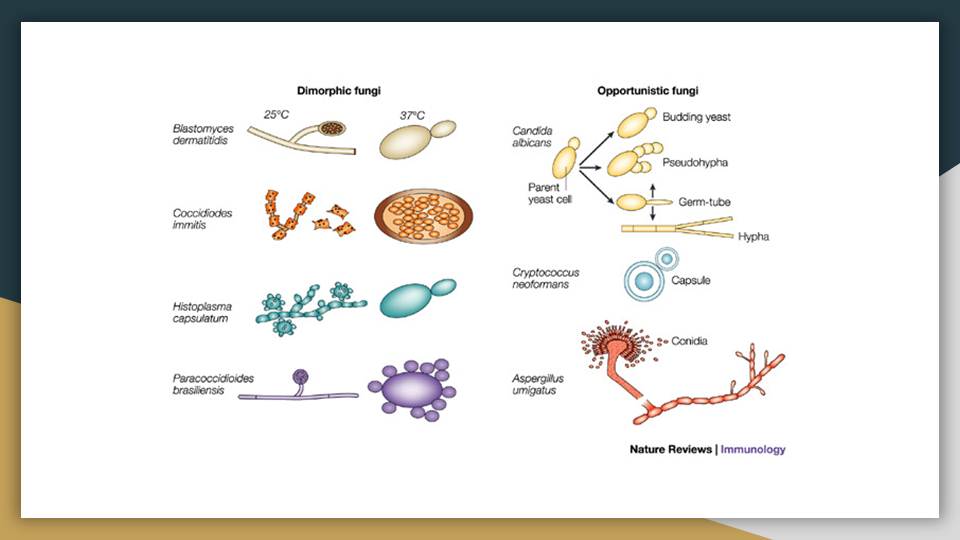
The Unique Dimorphism:
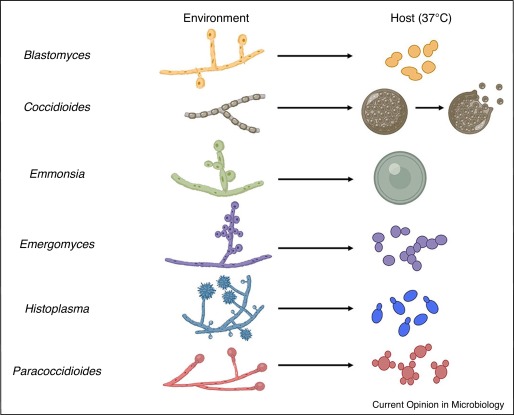
- Unlike Histoplasma and Blastomyces (which transition from mold to yeast), Coccidioides exhibits a unique dimorphism. It transitions between a mycelium (mold) in the cold environment and massive Spherules in the warm human tissue. It does NOT form a yeast phase!
- Both forms of growth are asexual, although comprehensive population genetic studies strongly suggest that a cryptic sexual phase does exist in nature to maintain genetic diversity.
III. The Fungal Life Cycle (Extremely High-Yield)
Understanding the life cycle is paramount for understanding transmission and histopathology.
The Saprobic Phase (25°C - Environment)
- Habitat: Alkaline, highly saline desert soil. Grows by apical extension with the formation of true septa. Maturation takes about one week. Mature, thin-walled mycelia undergo autolysis (programmed self-destruction) to provide nutrients as new young ones mature.
- Arthroconidia (The Infectious Spores): The mycelium fragments into highly durable, alternating, barrel-shaped spores called arthroconidia.
- Properties: They possess incredibly thick, hydrophobic walls, allowing them to remain viable in harsh, baking desert environments for years. They are highly prone to separation by physical disruption or even mild air turbulence, becoming airborne in a microscopic size (2-5 µm) perfectly capable of bypassing mucosal defenses and depositing deep into the human pulmonary alveoli.
The Parasitic Phase (37°C - Human Tissue)
- In the warm, moist lungs, the inhaled arthroconidia lose their thick hydrophobic outer walls.
- They remodel and swell into massive spherical cells called Spherules (which can grow to a gargantuan size, up to 100 μm in diameter).
- Endosporulation: Inside the Spherule, rapid nuclear division and cell multiplication occur. Septa transect the growing spherule into scores of subcompartments. Each subcompartment generates viable daughter cells called Endospores.
- Rupture: The outer spherule wall thins and ruptures after about 4 days, releasing hundreds of infectious endospores into the surrounding tissue. Each endospore forms a new spherule, creating an exponential infection loop.
The Spherule
If you see a massive, round sac completely packed with tiny, round dots (endospores) bursting open on a biopsy slide, it is definitive for Coccidioides. Think of a large piñata filled with microscopic candy breaking open inside the terminal bronchioles of the lungs.
IV. Epidemiology & Transmission
- Geography: Strictly endemic only to the alkaline soils of the Western Hemisphere (Southwestern USA, Northern Mexico, Central/South America). Nearly all cases exist within the north and south 40-degree latitudes (The "Lower Sonoran Life Zone").
- Seasonality: Within endemic regions, prevalence varies wildly with seasons. The fungus grows during heavy winter rains and subsequently dries and aerosolizes during the arid, windy summers.
- Dust Storms & Fomites (Extreme Outbreaks): Transport of arthroconidia—either in soil on fomites (archaeological equipment, contaminated cotton, military gear) or as the result of unusually severe weather events—has produced bizarre outbreaks.
Example: The massive "Haboob" dust storms in Arizona, or the 1994 Northridge earthquake in California, which caused landslides that aerosolized billions of spores, triggering an epidemic of Valley Fever hundreds of miles away in non-endemic zones. - Transmission Routes:
- Inhalation of arthroconidia: Accounts for 99.9% of all infections.
- Cutaneous inoculations: Exceptionally rare (e.g., a laboratory worker pricking a finger with a contaminated needle). Produces local lymphatic extension to regional lymph nodes and is typically self-limiting.
V. Pathogenesis & Histopathology
Pathogenesis (The Pathway of Infection):
- Inhalation of arthroconidia ➔ Deposition deep within the terminal bronchioles.
- Infective Dose: Only ONE single arthroconidium is required to initiate severe infection.
- The arthroconidium transforms into a spherule ➔ The fungus reacts with host complement, releasing potent chemokines that summon massive numbers of neutrophils ➔ Severe local inflammation ensues ➔ Formation of a local pulmonary lesion.
Dissemination (Escaping the Lungs):
- Fungal elements can move from the distal bronchiole into the lung parenchyma and gain entry into the vascular space.
- Endospores act as "Trojan Horses," hiding within macrophages to travel through the lymphatics to the bloodstream, creating highly lethal extrapulmonary sites of infection.
- Lymph node involvement classically includes the hilar, peritracheal, and cervical lymph nodes.
Histopathology: Acute vs. Chronic Responses
| Pathological State | Associated Fungal Stage | Inflammatory Infiltrate & Characteristics |
|---|---|---|
| Acute Inflammation | Active infections and rupturing spherules releasing endospores. | Intense influx of Neutrophils and Eosinophils. (Board Exam Note: Profound eosinophilia in the presence of a fungal pneumonia heavily and specifically points to Coccidioides!). |
| Chronic Inflammation | Arrested infections and mature, unruptured spherules. | Granulomatous lesions composed of lymphocytes, histiocytes, and multinucleated giant cells effectively walling off the intact spherules. |
VI. Host Defenses: The Immune Battlefield
1. Role of Innate Immunity:
- Includes Neutrophils (PMNs), Macrophages, and Natural Killer (NK) cells.
- Neutrophils are largely not fungicidal against Coccidioides.
- Macrophages & NK cells are fungicidal, but only against arthroconidia or young spherules. They physically cannot phagocytose or destroy massive, mature, thick-walled spherules.
- Conclusion: Innate responses serve to slow (rather than eliminate) fungal proliferation, forcing the infection into a subacute or chronic disease process while waiting for the adaptive immune system.
2. Role of T-Cells (Cellular Immunity):
- The ultimate survival and control of coccidioidomycosis depends entirely on T-lymphocytes (Th1 response).
- This is brutally evidenced by the increased severity and 100% mortality of naturally acquired infections in T-cell–deficient patients (e.g., untreated HIV/AIDS).
- In severe, uncontrolled disseminated cases, there is virtually no interferon-γ (IFN-γ) response to coccidioidal antigens, confirming an absent or broken Th1-type protective response.
3. Role of Antibody / B-Cell Responses:
- Coccidioidal infections give rise to a wide, massive variety of humoral (antibody) responses.
- Crucial Rule: Antibodies play absolutely NO ROLE in actual host defense against clearing Coccidioides species! High antibody titers actually correlate with worsening disease. They are only useful to the physician as a diagnostic and prognostic marker.
VII. Clinical Manifestations
The manifestations of most early coccidioidal infections overlap substantially with those of other respiratory infections (e.g., flu, community-acquired bacterial pneumonia), leading to frequent misdiagnosis.
- Early Respiratory Infection: 60% of patients are asymptomatic. The rest experience constitutional symptoms including profound weakness, high fever, general malaise, night sweats, and severe weight loss. Commonly accompanied by allergic dermatologic reactions like erythema nodosum (painful red nodules on the shins) and erythema multiforme, prompting the historic term "Desert Rheumatism."
- Pulmonary Disease: Can chronically progress to massive pulmonary nodules, thin-walled cavities (which can rupture and cause pneumothorax), or chronic fibrocavitary pneumonia mimicking Tuberculosis.
- Extrapulmonary Dissemination: The fungus escapes the lungs and attacks the meninges (Coccidioidal meningitis is 100% fatal without life-long therapy), bones (osteomyelitis), joints, and skin (warty, ulcerating lesions). Highly dangerous. Dissemination is statistically much higher in pregnant women (due to altered hormones) and individuals of African or Filipino descent.
VIII. Laboratory Diagnosis
Specific laboratory testing is required to establish a definitive diagnosis. It is established in three main ways: identifying the organism directly, detecting circulating antibodies, or detecting delayed-type hypersensitivity (skin testing).
- Microscopy & Stains:
- Direct wet preparations, KOH preps, Calcofluor white fluorescent staining, and Gram staining.
- Cytology/Histology stains: H&E, Gomori methenamine silver (GMS) staining, and PAS (Periodic acid–Schiff). Pathologists look for the classic 20-100 µm large spherules containing endospores.
- Culture (SEVERE DANGER!):
- Grows easily and well on most mycologic/bacteriologic media after 5 to 7 days of aerobic incubation. Appears as a fluffy, white, nonpigmented mold.
- Safety Warning: Culture containers must ONLY be opened inside a certified BSL-3 biocontainment cabinet! Opening a standard petri dish of Coccidioides on an open laboratory bench will instantly release millions of invisible arthroconidia and fatally infect the entire laboratory staff.
- Identification is legally confirmed via specific exoantigen detection in a fungal extract or specific rRNA sequencing using a DNA probe.
- Serologic Testing:
- The most frequent, safest means of diagnosing primary infections. It is highly specific for active infection.
- Clinical Trap: A negative serologic test early on never excludes infection! Performing repeated, serial tests over the course of 2 months heavily increases diagnostic sensitivity.
- Common Tests: Tube Precipitin (Detects IgM - indicates early/acute infection), Complement-Fixing Antibodies (Detects IgG - tracks disease severity, dissemination, and CSF involvement in meningitis), Immunodiffusion, ELISA, and Latex Agglutination Tests.
- Antigen & Molecular Detection:
- Antigenemia (fungal proteins in the blood/urine) may occur with early or chronic immunocompromised infections.
- PCR (Polymerase Chain Reaction): Detects C. immitis-specific nucleic acid sequences directly in patient sputum or tissue specimens, entirely bypassing the extreme biological danger of having to culture the mold.
IX. Therapy & Prevention
Management Components:
- Assessment of the need for intervention (many mild, immunocompetent cases self-resolve with purely supportive care).
- Selection of potent systemic antifungal agents.
- Choice of surgical procedures for the aggressive debridement and reconstruction of destructive lesions (e.g., lobectomy for massive lung cavitations or scraping of necrotic bone lesions).
- Amphotericin B: The intravenous "heavy-hitter." It binds ergosterol and tears holes in the fungal membrane. Reserved for disseminated, immediately life-threatening, or severe respiratory disease. Extremely nephrotoxic.
- Azoles: Ketoconazole, Fluconazole, and Itraconazole. Used for mild/moderate disease, chronic bone infections, or as oral step-down therapy. Clinical Note: Coccidioidal Meningitis requires life-long, daily high-dose Fluconazole because it crosses the blood-brain barrier effectively.
- Voriconazole & Caspofungin: Used as salvage therapy for refractory cases.
- Nikkomycin Z:
Physiology Expansion: This is a highly specific, experimental anti-fungal that directly targets chitin synthase, the enzyme required to build the rigid fungal cell wall. Because human cells do not possess chitin or chitin synthase, it is highly selective and boasts an incredibly low toxicity profile compared to Amphotericin B!
Prevention & Vaccines:
- Lifelong, robust immunity naturally develops in almost all persons who are infected and subsequently recover.
- A formalin-killed, whole-cell spherule vaccine was created historically but failed spectacularly in human trials. It induced a great deal of severe, painful local inflammation and sterile abscesses at the injection site.
- Future prevention relies on developing purified or recombinant protein antigens, which might circumvent this toxic limitation and safely induce Th1 memory.
❓ Applied Clinical Question: The Archaeologist
Case: An archaeologist excavating Native American ruins in the deep deserts of Arizona develops a severe, progressive pneumonia with a high fever and painful red nodules on his shins. A complete blood count reveals marked eosinophilia. Serology is positive for high-titer complement-fixing (CF) IgG antibodies. What specific fungal structure is currently growing and replicating inside his lung tissue?
Answer: He inhaled environmental Arthroconidia from the Arizona dust, but the structure currently growing and destroying his lungs is the Spherule (filled with endospores). The geographic location (Southwestern US desert), the intense dust/soil exposure, the classic erythema nodosum, and the profound eosinophilia make Coccidioides the definitive and undeniable diagnosis.
Part 2: Paracoccidioides brasiliensis (South American Blastomycosis)
I. Introduction to Paracoccidioides
Paracoccidioides brasiliensis is the causative agent of Paracoccidioidomycosis (formerly known historically as South American Blastomycosis). It is clinically recognized as the single most important and prevalent endemic systemic fungal disease in Latin America.
- Disease Characteristics: It causes a chronic, systemic, and highly progressive granulomatous disease that is frequently fatal if left untreated.
- Primary Infection Site: The lungs (via inhalation of environmental propagules).
- Dissemination Pathways: Readily spreads via lymphohematogenous routes to mucous membranes, skin, the reticuloendothelial system (RES - spleen, liver, lymph nodes), and importantly, the adrenal glands.
Clinical Note: Massive fungal destruction of the adrenal cortex leads to secondary Addison's disease (adrenal insufficiency), presenting with severe hypotension, hyperkalemia, and generalized skin hyperpigmentation. - Epidemiology: Distinctly affects adult men who work in agriculture. Geographic distribution is strictly restricted to Latin America (Brazil, Colombia, Venezuela, Argentina), specifically concentrating in rural coffee and tobacco-growing regions characterized by mild temperatures, high constant humidity, and heavy annual precipitation.
Taxonomic Classification:
Kingdom: Fungi ➔ Phylum: Ascomycota ➔ Class: Eurotiomycetes ➔ Order: Onygenales ➔ Family: Ajellomycetaceae ➔ Genus: Paracoccidioides.
The 15:1 Male-to-Female Ratio
Epidemiological data notes a massive, undeniable gender disparity: the disease affects men compared to women at a staggering ratio of 15:1 to 70:1 in adults (>30 years of age). Why?
It was discovered that female sex hormones—specifically 17-beta-estradiol—directly bind to a specialized cytosolic hormone-binding protein within the fungus. This binding completely inhibits the transition of the inhaled environmental mycelium into the pathogenic, disease-causing yeast cells at 37°C! Adult females in rural areas inhale the exact same spores from the environment just as frequently as men do, but the estrogen circulating in their bodies prevents the fungus from transforming into its parasitic phase, granting women profound natural immunity.
(Interestingly, in prepubescent children who lack high estrogen, the male-to-female ratio is 1:1).
II. The Organism: Morphology & Virulence
Morphology & Growth Characteristics:
It is a true thermally dimorphic fungus. Currently, only the anamorph (asexual) characteristics are widely known and studied.
Parasitic Phase (37°C)
- Found natively in cultures incubated at 37°C, as well as heavily in human tissues, sputum, and purulent exudates.
- Colonies grow extremely slowly (about 10 to 20 days) and appear as soft, cream-colored, heavily wrinkled cerebriform (brain-like) mounds.
- Microscopic Appearance: Uniquely large cells (4 to 40 μm) featuring a thick, translucent, double-contoured cell wall and prominent intracytoplasmic lipid globules.
- Reproduction: Multiplies by multiple, simultaneous budding. The blastoconidia (daughter cells) are small (4 to 6 μm) and remain rigidly connected to the massive central mother cell by short, narrow cytoplasmic bridges.
Saprobic Phase (26°C - Environment)
- A very slow-growing mold (takes 20 to 30 days to mature).
- On solid agar media, it produces sterile, white, cottony aerial mycelia that adhere aggressively to the agar surface; the area directly beneath the colony turns a brownish-yellow.
- Microscopically contains tough chlamydospores and extremely thin septate hyphae. On nutrient-deprived media with reduced carbohydrate content, it produces the true infectious particles: arthroconidia.
The "Pilot Wheel" or "Mariner's Wheel"
Under the microscope, observing a massive central mother yeast cell entirely surrounded by a 360-degree crown of multiple tiny budding daughter cells—all attached by very thin, delicate cytoplasmic bridges—looks exactly like the wooden steering wheel of an old pirate ship. On board exams and in clinical microscopy, describing a "Pilot Wheel" or "Mariner's Wheel" appearance instantly equals Paracoccidioides brasiliensis.
Ecology & Virulence Factors:
- Reservoirs/Habitat: Acidic, humid soils, commercial dog chow, penguin feces, and specifically the nine-banded armadillo (Dasypus novemcinctus), which is highly susceptible and acts as an environmental amplifier.
- Transmission: Strict inhalation of fungal spores from disturbed soil (e.g., harvesting coffee beans). There is absolutely no human-to-human transmission. Incubation can involve extraordinarily long periods of clinical latency (the fungus goes dormant and can reactivate up to 30 years after leaving the endemic zone!).
- Virulence Characteristics:
- gp43: An incredibly potent, 43-kDa immunodominant glycoprotein antigen excreted by the yeast. It serves heavily in laboratory diagnosis, possesses powerful cellular adhesive functions (facilitating tissue invasion), and acts as a direct immunosuppressant against host macrophages.
- Morphologic transition capabilities (the strict ability to switch from mold to yeast at body temperature).
- Adherence capabilities, destructive proteolytic enzymes, and immune-evading Melanin production within the cell wall.
III. Pathogenesis & Host Defenses
1. Innate Immunity:
- Involves PMNs (neutrophils), alveolar macrophages (MQS), NK cells, the complement cascade, and proinflammatory cytokines.
- These early innate defenses severely hinder initial fungal multiplication but are ultimately biologically unable to destroy the robust yeast on their own.
2. Adaptive Immunity & The Critical Th1/Th2 Balance:
- Antibodies (B-cell responses): Bear no direct role in biological protection. In fact, severe, widespread, uncontrolled disease is characterized by massive, useless antibody production (hypergammaglobulinemia).
- Protection: Relies entirely on robust Granuloma formation (the most effective biologic defense weapon against P. brasiliensis). Granuloma formation absolutely requires functional T lymphocytes (CD4 & CD8), fully activated macrophages, and high levels of Th1 cytokines (IFN-γ and IL-12).
- The Macrophage Mechanism: Macrophages, when properly activated by IFN-γ, ingest and completely obliterate the conidia/yeasts via the powerful L-arginine–nitric oxide (oxidative burst) pathway.
- The Danger of Th2: Th2 cytokines (IL-4, IL-5, IL-10, TGF-β) actively and chemically interfere with macrophage function, switching off their killing mechanisms. If a patient genetically or environmentally mounts a Th2 response instead of a protective Th1 response, the fungus spreads uncontrollably, leading to the highly fatal juvenile form of the disease.
IV. Clinical Manifestations
Infection almost always begins as a subclinical (asymptomatic) pulmonary process. Evidence of past infection includes a reactive skin test, circulating anti-GP43 antibodies, or small residual fibrotic lung lesions. Overt disease, when it strikes, is highly polymorphic, severe, and progressive, initially presenting with constitutional symptoms (profound weakness, daily fever, malaise, severe weight loss/cachexia).
The Three Main Clinical Presentations:
- Chronic Adult Form (Accounts for 90% of cases):
- Presents as unifocal or multifocal disease. Unifocal disease features intermediate, somewhat competent immune responses (lymphoproliferative).
- Nearly always secondary to endogenous reactivation years (or decades) after the initial respiratory exposure.
- Triggers for reactivation: Aging, immunosuppression, debilitating concomitant disease, chronic severe alcoholism, malnutrition, and heavy tobacco smoking.
- Targets: Mainly destroys the lungs (causing bilateral, patchy infiltrates and severe fibrosis), with secondary horrific lesions breaking out on mucous membranes.
Clinical Note: Mucocutaneous lesions are pathognomonic—patients develop painfully ulcerated, mulberry-like granulomatous stomatitis in the mouth/gums, leading to spontaneous tooth loss, dysphagia (inability to swallow), and destruction of the hard palate and nasal septum. It also attacks the RES, skin, and adrenal glands.
- Juvenile Form (Accounts for <15% of cases):
- Acute/subacute and significantly more severe and aggressive. Represents rapid progression immediately after a recent, heavy environmental exposure.
- Targets: Massive, devastating involvement of the reticuloendothelial system (leading to massive hypertrophy of lymph nodes with purulent drainage, and severe hepatosplenomegaly).
- Presents with minimal respiratory complaints but carries a horrific prognosis and high mortality rate.
- Immune Profile: Nonreactive (anergic) skin tests, wildly high but useless detectable antibodies, severely depressed cellular lymphoproliferative response to gp43, and a heavy, flawed Th2 cytokine pattern (low IFN-γ; wildly high IL-4, IL-5, IL-10; completely absent IL-12).
- Residual Form: Dense fibrotic scarring of previously active lesions. This scarring can cause severe sequelae, such as permanent tracheal stenosis (narrowing of the windpipe), pulmonary cor pulmonale (right-sided heart failure), and permanent adrenal insufficiency.
V. Diagnosis & Treatment of Paracoccidioidomycosis
Differential Diagnosis (DDx):
Because the presentation is so variable, it is frequently misdiagnosed as Tuberculosis, Neoplastic disorders (lymphoma/oral squamous cell carcinoma), Histoplasmosis, Mucocutaneous Leishmaniasis, Leprosy, and Syphilis. Only the laboratory is capable of establishing the correct, definitive diagnosis.
Laboratory Diagnosis:
- Direct Examination: Sputum, scraping of oral ulcers, exudates, and lymph node pus. Wet mounts (KOH) or calcofluor preps instantly reveal the classic multiple-budding "Pilot Wheel" yeast.
- Histology: Tissue biopsy of lymph nodes or skin. Gomori's methenamine silver (GMS) stain is the most recommended. Shows intense mixed inflammatory reactions (suppurative and granulomatous) centered heavily on the large yeast cells.
- Culture: Sabouraud-dextrose agar supplemented with antibacterial agents and cycloheximide to prevent overgrowth. Requires a massive 6 weeks of incubation at room temperature and 37°C. A positive culture equals active, undeniable infection.
- Serologic Tests: Used heavily for both initial diagnosis and long-term follow-up. Detects Ig G, M, and E directed specifically against gp43, pb27, and HS proteins.
- Agar gel immunodiffusion: The easiest and best method; 90% sensitive and highly specific.
- Complement Fixation: Highly cumbersome, and severely cross-reacts with Histoplasma capsulatum, creating false positives.
- Antigen Tests: Monoclonal antibody techniques detect circulating fungal antigens with 60% sensitivity in serum/CSF/urine. Clinical Note: Decreasing antigen titers strongly and definitively correlate with positive clinical improvement and drug efficacy!
- Note on Skin Tests: Paracoccidioidin intradermal skin testing is completely unreliable for active diagnosis. 35-50% of active severe cases are nonreactive (anergic due to immune exhaustion), and the antigen heavily cross-reacts with histoplasmin.
Treatment Protocols:
Therapy must be maintained for prolonged periods (months to years) to prevent devastating relapses.
- The Sulfa Exception: Paracoccidioidomycosis is uniquely the ONLY systemic mycosis highly amenable to treatment with inexpensive sulfa drugs! Sulfonamides (e.g., Trimethoprim-sulfamethoxazole/Cotrimoxazole, sulfadimethoxine) are highly effective and widely used in rural Latin America.
- Primary Azole Agents: Itraconazole (the modern drug of choice due to high efficacy and short duration of 6 months) and Ketoconazole.
- Severe Disease: Intravenous Amphotericin B is mandated for rapidly progressive, life-threatening juvenile forms or severe pulmonary compromise, followed by oral step-down therapy.
- Newer Therapies: Posaconazole (a powerful Triazole with excellent salvage rates) and Terbinafine.
- Avoid: Fluconazole is strictly NOT recommended due to inexplicably high clinical failure rates (relapses) and the necessity for massive, poorly tolerated doses to achieve any response.
- Holistic Management: Treatment directed exclusively at the fungus may fail. Correction of the patient's underlying debilitating disease (e.g., vastly improved high-protein diet, forced bed rest, correction of severe anemia, stopping smoking/alcohol) and immunomodulant adjuvant therapy (like recombinant IFN-γ) are absolutely necessary for survival in severe cases.
❓ Applied Clinical Case: The Coffee Farmer
Case: A 54-year-old male coffee plantation worker from rural Brazil presents to the clinic complaining of extreme weight loss, a chronic productive cough, and painful, bleeding, ulcerated lesions throughout his gums that have caused two of his teeth to fall out. His blood pressure is unusually low (90/60 mmHg), and his skin appears abnormally hyperpigmented. A biopsy of the oral mucosa reveals large yeast cells with multiple, narrow-based buds forming a "mariner's wheel" structure.
Clinical Correlation: What is the diagnosis, what explains his gender susceptibility, and what explains his low blood pressure/hyperpigmentation?
Answer:
1. Diagnosis: Chronic Adult Paracoccidioidomycosis (South American Blastomycosis).
2. Gender Susceptibility: As an adult male, he lacks circulating estrogen (17-beta-estradiol), which normally binds to the fungus and prevents the infectious mold from transforming into the invasive yeast at body temperature.
3. Blood Pressure/Skin: The fungus has a high predilection for disseminating to and destroying the adrenal glands. His low blood pressure and hyperpigmentation are classic signs of Addison's disease (primary adrenal insufficiency) caused by fungal infiltration.
List of References
- Bennett, J. E., Dolin, R., & Blaser, M. J. (2019). Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases (9th ed.). Elsevier.
- Murray, P. R., Rosenthal, K. S., & Pfaller, M. A. (2020). Medical Microbiology (9th ed.). Elsevier.
- Kumar, V., Abbas, A. K., & Aster, J. C. (2020). Robbins & Cotran Pathologic Basis of Disease (10th ed.). Elsevier.
- Kauffman, C. A., Pappas, P. G., Sobel, J. D., & Dismukes, W. E. (2011). Essentials of Clinical Mycology (2nd ed.). Springer.
- Galgiani, J. N., Ampel, N. M., Blair, J. E., et al. (2016). "2016 Infectious Diseases Society of America (IDSA) Clinical Practice Guideline for the Treatment of Coccidioidomycosis." Clinical Infectious Diseases, 63(6), e112-e146.
- Shikanai-Yasuda, M. A., Mendes, R. P., Colombo, A. L., et al. (2017). "Brazilian guidelines for the clinical management of paracoccidioidomycosis." Revista da Sociedade Brasileira de Medicina Tropical, 50(5), 715-740.
Quick Quiz
Intro to Mycology Quiz
Microbiology - mobile-friendly and focused practice.
Privacy: Your details are used only for quiz tracking and certificates.
Intro to Mycology Quiz
Microbiology
Preparing questions...
Choose your answer and keep your streak alive.
Great effort.
Here is your quick performance summary.
Coccidioides & Paracoccidioides brasiliensis Read More »