

Table of Contents
ToggleSickle Cell Disease/Sickle Cell Anaemia
Sickle cell disease is an inherited red-blood cell disorder which causes the body to produce abnormally shaped red blood cells.
Sickle cell disease is inherited as an autosomal recessive trait. Normal Hb A gets replaced with Abnormal Hb S.
Children with this disorder have atypical haemoglobin molecules called haemoglobin S which can distort red blood cells into a sickle or crescent shape.

Red blood cells with normal hemoglobin are smooth, disk-shaped, and flexible, like doughnuts without holes. They can move through the blood vessels easily.
Cells with sickle cell hemoglobin are stiff and sticky. When they lose their oxygen, they form into the shape of a sickle or crescent, like the letter C.
These cells stick together and can’t easily move through the blood vessels. This can block small blood vessels and the movement of healthy, normal oxygen-carrying blood. The blockage can cause pain

Classification of sickle cell disease
Disease is broadly classified into;
1. Sickle Cell Anaemia (Homozygous): Are patients whose Red blood cells only contain abnormal beta chains leading to HbSS (SS). These patients are said to have sickle-cell anaemia and they have S+S of Sickle cell disease. Individuals with sickle cell anaemia inherit two copies of the faulty haemoglobin gene, one from each parent. This is denoted as HbSS or SS. Other names: HbSS, SS disease, Haemoglobin S.
2. Sickle Cell Trait (Heterozygous): Patients whose Red blood cells contain a mixture of normal beta chains of HbA and abnormal beta chains of HbS. Thus patients have both HbA and HbS (HbAS). Individuals with sickle cell trait inherit one copy of the normal haemoglobin gene and one copy of the faulty haemoglobin gene. This is denoted as HbAS. People with sickle cell trait are usually asymptomatic, meaning they don’t experience the typical symptoms of SCD. They are carriers of the faulty gene and can pass it on to their children.
To understand Homozygous and Heterozygous,
SCD (Sickle Cell Disease): Think of this as a house built with a faulty instruction manual. The manual has instructions for building strong, healthy red blood cells (the “bricks” of your blood), but the instructions are messed up. This leads to problems with the shape and function of red blood cells, causing sickle cell disease.
Autosomal: This refers to the chromosomes that determine most of your traits, except for sex (male or female). Imagine these chromosomes like the foundation of your house.
Heterozygous: You have two copies of each autosomal chromosome, one from each parent. Imagine you received an instruction manual with good instructions from your mom and a manual with a faulty set from your dad. This means you have a good copy and a faulty copy of the gene that causes sickle cell disease. You are a “carrier” of the faulty gene, but you don’t have SCD.
Homozygous: You received the same instruction manual from both parents. There are two possibilities:
- Homozygous dominant: You received two good instruction manuals (from both parents). Your house is built strong and healthy, you don’t have SCD.
- Homozygous recessive: You received two faulty instruction manuals (from both parents). Your house has serious problems, you have SCD.
Recessive: A recessive gene only causes a disease when you have two faulty copies (like in the homozygous recessive case). Think of it as needing two faulty instruction manuals to build a house with problems.
Dominant: A dominant gene always causes a disease, even if you only have one faulty copy (like in the heterozygous case). Imagine the faulty instruction manual overrides the good one.
Summary:
- SCD: A faulty instruction manual leads to problems with red blood cells.
- Autosomal: The chromosomes that determine most traits (the house’s foundation).
- Heterozygous: You have one good and one faulty copy of a gene (one good and one faulty instruction manual).
- Homozygous: You have two identical copies of a gene (two good or two faulty instruction manuals).
- Recessive: You need two faulty copies to express the disease (two faulty instruction manuals to build a bad house).
- Dominant: You only need one faulty copy to express the disease (one faulty instruction manual is enough to build a bad house).
- Red Blood Cells: These cells carry oxygen throughout the body.
- Haemoglobin: A protein within red blood cells that binds to oxygen.
- Haemoglobin Gene: A gene located on chromosome 11 that provides instructions for making haemoglobin.
Possibility of Sickle cell Disease
Problems in sickle cell disease begin around 5 to 6 months of age. Sickle-cell disease occurs when a person inherits two abnormal copies of the haemoglobin gene, one from each parent. This gene occurs in chromosome 11.
| Type of Gene | Normal | Trait | Disease |
|---|---|---|---|
| One Parent with Trait | 50% | 50% | 0% |
| Both Parents with Trait | 25% | 50% | 25% |
| One Parent with Disease | 50% | 50% | 50% |
| Both Parents have Disease | 0% | 0% | 100% |
Cause of Sickle Cell Disease
- It is caused by a defect in beta chains where a given amino acid is replaced by another (Substitution of valine for glutamic acid) at position 6 of the chain.
- This change creates abnormal hemoglobin called HbS.
Sickle cell disease is caused by a genetic mutation in the gene that produces haemoglobin, a protein in red blood cells that carries oxygen.
- Normal Haemoglobin: Normal haemoglobin is made up of two alpha chains and two beta chains, denoted as HbA.
- Sickle Cell Haemoglobin: In sickle cell disease, there’s a single point mutation in the beta chain of haemoglobin, replacing a glutamic acid with valine, at position 6 of the chain.This mutated haemoglobin is called HbS.
Pathophysiology of Sickle Cell Disease.
Normally each haemoglobin molecule consists of four molecules of haem folate into one molecule of globin.
But in sickle cell disease this is altered and cells become sickle shaped, glutamine is replaced by valine. The sickle cells elongate under conditions of lower oxygen concentration, Acidosis takes place and dehydration.
When red blood cells (RBCs) containing homozygous HbS are exposed to deoxygenated conditions, the sickling process begins. This distorts the membranes of red blood cells. The cell becomes easily entangled leading to blood viscosity, vessel occlusion and tissue necrosis.
These cells fail to return to normal shape when normal oxygen tension is restored. As a result, these rigid blood cells are unable to deform as they pass through narrow capillaries, leading to vessel occlusion and ischemia. The actual anaemia of the illness is caused by haemolysis, the destruction of the red cells, because of their shape.
Although the bone marrow attempts to compensate by creating new red cells, it does not match the rate of destruction. Healthy red blood cells usually function for 90–120 days, but sickled cells only last 10–20 days. Increased sequestration of Red blood cells in the spleen also cause anaemia
Clinical Presentation of SCD
Children are rarely symptomatic until late in the first years of life related to increased amounts of fetal haemoglobin being cleared from blood. The severity of symptoms can vary from person to person. Sickle-cell disease may lead to various acute and chronic complications, several of which have a high mortality rate.
- Painful swelling of hands and feet (Hand-foot syndrome): This is a common presentation in children, caused by vaso-occlusive crisis in the small blood vessels of the hands and feet.
- Pain crisis (sickle crisis): This is a major complication characterized by intense pain due to blocked blood flow to a specific area of the body and can last for days or weeks. Pain in the chest, abdomen, limbs, and joints.
- Anaemia: A consistent feature, as the lifespan of sickle red blood cells is shortened. This leads to fatigue, weakness, and paleness.
- Jaundice: Caused by the breakdown of red blood cells, leading to a yellowish discoloration of the skin and eyes.
- Haemoglobin levels: Usually low, ranging from 6 g/dL to 9 g/dL, indicating the severity of anaemia.
- Shortness of breath: Caused by complications like pneumonia, acute chest syndrome, and pulmonary hypertension.
- Fatigue and weakness: A common symptom due to the low oxygen levels caused by anaemia.
- Priapism: A painful erection lasting for hours or days, caused by blocked blood flow in the penis. If not promptly treated, it can lead to impotence.
- Abdominal swelling and pain: Often associated with spleen enlargement (splenomegaly) or blockages in the blood vessels supplying the intestines.
- Unusual headache: May be a sign of stroke, as sickled cells can block blood flow to the brain.
- Loss of appetite: A common symptom associated with anaemia and pain.
- Irritability: Can be a response to pain, fatigue, or other symptoms.
- Bossing of the bones of the skull: Indicates active erythropoiesis (red blood cell production) to compensate for the loss of sickle cells.
- Intercurrent infections: Patients with sickle cell disease are more susceptible to infections like pneumonia, acute respiratory infections, and malaria, often complicated by severe anaemia.
- Splenomegaly: Enlarged spleen, common in younger children, but often shrinks in older children due to splenic infarction.
- Growth retardation: Can occur due to chronic illness, pain, and infections.
- Stroke: A serious complication resulting from blocked blood flow to the brain, leading to brain damage.
Newborns: May present with jaundice, delayed cord clamping, and possible failure to thrive.
Children:
- Dactylitis (Hand-foot Syndrome): Painful swelling of hands and feet due to vaso-occlusive crisis.
- Splenomegaly: Often present in young children, but can be absent in older children due to splenic infarction (damage).
- Delayed growth and development are common due to recurrent infections and pain crises.
- Delayed puberty: Can be a feature, especially in males.
Adults:
- Chronic pain is a defining feature, often with unpredictable patterns.
- Pulmonary complications: Pulmonary hypertension, acute chest syndrome, and pneumonia are frequent issues.
- Osteonecrosis: Damage to bone due to lack of blood flow.
- Avascular necrosis: Can affect bones, especially hips and shoulders.
- Chronic kidney disease: Can develop over time due to repeated damage to the kidneys.
Chronic Symptoms:
- Jaundice: Yellowing of the skin and whites of the eyes due to the breakdown of red blood cells.
- Gallstones: Formation of stones in the gallbladder, often caused by a build-up of bilirubin from red blood cell breakdown.
- Progressive kidney impairment: Damaged blood vessels in the kidneys can lead to reduced kidney function over time.
- Growth retardation: Slower growth of long bones and skeletal deformities, particularly in the spine, can occur.
- Delayed puberty: The chronic illness can delay the onset of puberty.
- Chronic painful leg ulcers: Related to chronic anaemia and poor blood flow to the extremities.
- Decreased lifespan: While advancements in medical care have improved life expectancy, individuals with sickle cell disease still have a shortened lifespan compared to the general population.
- Altered body structures: These include “bossing” of the skull (abnormal thickening of the skull bones), as well as septic necrosis (bone death due to infection) in the femur (thigh bone) and head of the humerus (upper arm bone).
Sickle-cell crisis
Sickle cell crisis is pain that can begin suddenly and lasts several hours to several days.
The terms “sickle-cell crisis” or “sickling crisis” may be used to describe several independent acute conditions occurring in patients with Sickle Cell Disease. It happens when sickled red blood cells block small blood vessels that carry blood to bones. Children may present with pain in the back, knees, legs, arms, chest or stomach. The pain can be throbbing, sharp, dull or stabbing.
Types of Sickle Cell Crisis.
(i) Vaso-occlusive Crisis: This is the most common form of crisis. Small blood vessels are occluded by the sickle cells causing distal ischemia and infarction, leading to pain, swelling, and inflammation.
- Symptoms: Intense pain in the bones, joints, abdomen, chest, or head. Other symptoms may include fever, fatigue, and shortness of breath.
- Extremities. Bone destruction leading to osteoporosis or ischaemic necrosis.
- Foot and hand syndrome due to aseptic infarction of metacarpals and metatarsals causing swelling and pains often this is seen in infants and toddlers.
- Triggers: Dehydration, infection, cold weather, high altitude, and strenuous physical activity.
- Treatment: Pain management with analgesics, intravenous fluids, and blood transfusions in severe cases.
(ii) Splenic sequestration Crisis: Large amounts of blood become pooled to the spleen, leading to a decrease in blood volume and blood pressure. The spleen becomes massively enlarged.
- Symptoms: Abdominal pain, swelling, fever, and shock. Great decrease in Red blood cells mass occurs within hours. Signs of circulatory collapse develop rapidly.
- This is the most frequent cause of death in infants with sickle cell disease.
- Treatment: Immediate medical attention with intravenous fluids, blood transfusions, and sometimes splenectomy.
(iii) Aplastic Crisis: The bone marrow ceases to produce RBCs. A sudden drop in red blood cell production, leading to severe anaemia and worsening of symptoms. There will be low blood cell circulation in blood hence anaemia.
- Cause: Usually triggered by viral infections like parvovirus B19. Folic acid deficiency and Ingestion of bone marrow toxins (eg, phenylbutazone).
- Symptoms: Fatigue, weakness, pallor, and shortness of breath.
- Treatment: Blood transfusions to increase red blood cell count.
(iv) Haemolytic Crisis: Hemolytic crisis occurs when large numbers of red blood cells are destroyed over a short time. The loss of red blood cells occurs much faster than the body can produce new red blood cells.
- Cause: Often triggered by infections.
- Symptoms: Fatigue, pallor, jaundice, and dark urine.
- Treatment: Blood transfusions and treatment of underlying infections.
Causes of hemolysis include:
- A lack of certain proteins inside red blood cells
- Autoimmune diseases
- Certain infections
- Defects in the haemoglobin molecules inside red blood cells
- Defects of the proteins that make up the internal framework of red blood cells
- Side effects of certain medicines
- Reactions to blood transfusions.
(v) Acute chest syndrome. This occurs in the chest, when sickled red blood cells block blood flow to the lungs, leading to inflammation and damage. This can be life-threatening. It often occurs suddenly, when the body is under stress from infection, fever, or dehydration.
- Symptoms: Chest pain, fever, shortness of breath, cough, and rapid breathing.
- Treatment: Oxygen therapy, antibiotics, pain management, and sometimes mechanical ventilation.

Precipitating Factors of Sickle Cell Crisis
Sickle cell crises are painful episodes that occur when sickle red blood cells block blood flow in the body. These crises can be triggered by various factors, including:
Environmental and Physiological Factors:
- Dehydration: Lack of fluids can thicken the blood, making it harder for sickle cells to flow through small blood vessels.
- Infection: Infections can increase the body’s demand for oxygen, putting stress on already compromised red blood cells.
- Trauma: Injury, including even minor cuts or bruises, can lead to localized blood clotting and trigger a crisis.
- Extreme Temperature Fluctuations: Both extreme heat and cold can constrict blood vessels and lead to blockage.
- High Altitude: The thinner air at high altitudes can lead to oxygen deprivation, increasing the likelihood of sickling.
- Hypoxia: Low oxygen levels in the blood, from any cause, can trigger sickling.
- Acidosis: Increased acidity in the blood can also contribute to sickling.
Lifestyle and Emotional Factors:
- Strenuous Physical Exercise: Intense physical activity can increase the body’s demand for oxygen and contribute to sickling.
- Extreme Fatigue: Prolonged exhaustion weakens the body’s ability to fight off crises.
- Extreme Exertion: Similar to intense exercise, any extreme physical effort can trigger a crisis.
- Emotional Stress: Stress hormones can constrict blood vessels and increase the likelihood of sickling.
Other Contributing Factors:
- Pregnancy: The increased blood volume and hormonal changes during pregnancy can make women more susceptible to crises.
- Asthma: The inflammatory response in asthma can trigger sickle cell crises.
- Anxiety: Similar to stress, anxiety can constrict blood vessels and increase the risk of a crisis.
- Dehydration.
- Infection.
- Trauma.
- Strainous Physical exercises.
- Extreme fatigue.
- Extreme exertion
- Severe cold that constricts peripheral vessels
- Fever Excessive exercise
- Hypoxia.
- Acidosis.
- Extreme temperature
- High attitude
- Emotional stress
- Pregnancy
- Asthma
- Anxiety
- Abrupt changes in temperature
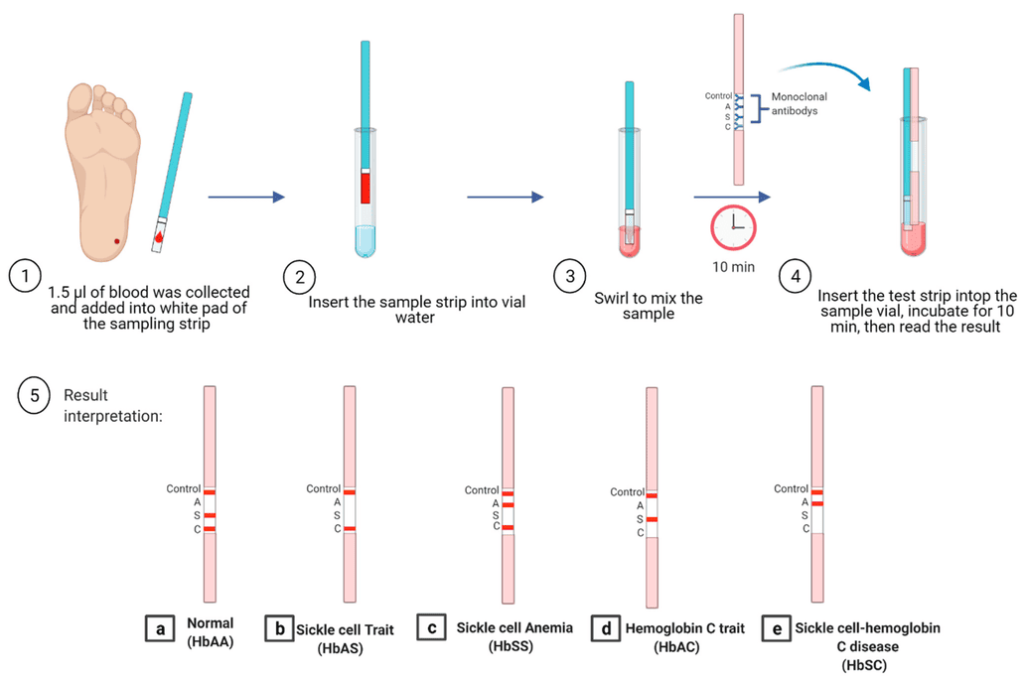
Diagnosis and Investigations:
- Family history: A strong family history of sickle cell disease is a big indicator.
- Full blood count and peripheral film: The blood test may show leukocytosis (increased white blood cell count) due to bacterial infection and reveal the presence of sickle cells.
- Haemoglobin estimation: Will reveals a low haemoglobin level (6-8 g/dL) with a high reticulocyte count (10-20%), indicating the body’s attempt to compensate for the loss of red blood cells.
- Sickling test: This simple test, done by finger or heel prick, observes a drop of blood under a microscope after removing oxygen. Sickle-shaped cells are indicative of the disease. However, it doesn’t distinguish between the trait and the disease or other sickle haemoglobin opathies.
- Haemoglobin electrophoresis: This more definitive test involves separating different types of haemoglobin through an electric current. It identifies the presence and amount of HbS (sickle haemoglobin), providing a definitive diagnosis for both the trait and the disease.
- Sickledex test: A rapid screening test for detecting the presence of HbS in the blood.
- Peripheral blood smear: Examines a blood sample under a microscope to identify sickle cells and reticulocytes.
- Urinalysis: Analyzes urine for signs of kidney damage.
- Liver and renal function tests: Assess the function of the liver and kidneys.
- Chest radiography: Used to diagnose Acute Chest Syndrome.
- Abdominal ultrasound: Can help detect problems in the abdomen, such as a mesenteric crisis (blockage of blood vessels in the intestines).
- Sickling test (emergency screening): Can be performed before surgery to identify individuals with sickle cell disease.
Differential Diagnosis
- Acute anaemia
- Carotid-Cavernous Fistula (CCF)
- haemoglobin C Disease
- Hemolytic Anaemia
- Osteomyelitis in Emergency Medicine
- Pulmonary Embolism (PE)
- Rheumatoid Arthritis Hand Imaging
- Septic Arthritis
Management of Sickle Cell Disease.
Management is according to the type of crisis .
Aims of Management
- Avoiding pain episodes.
- Relieving symptoms.
- Preventing complications.
- Acute painful attacks require supportive therapy with intravenous fluids, oxygen, antibiotics and adequate analgesia.
- Crises can be extremely painful and usually require narcotic analgesia. Morphine is the drug of choice. Milder pain can sometimes be relieved by codeine, paracetamol and NSAIDs.
- Oxygen Therapy: Supplementary oxygen is provided to address hypoxia and alleviate symptoms.
- Prophylaxis is with penicillin twice daily, up to 5 years of age due to the immature immune system that makes them more prone to early childhood illnesses is recommended and vaccination with polyvalent pneumococcal and Haemophilus influenzae type B vaccine .
- Hydration: Drinking plenty of fluids is essential to prevent dehydration and improve blood flow.
- Blood Transfusions: Regular transfusions are used to increase haemoglobin levels and reduce the frequency of crises. Transfusions should be given for heart failure, strokes, acute chest syndrome, acute splenic sequestration and aplastic crises.
- Anaemia Transfusions should only be given for clear indications.
- Patients with steady state anaemia, those having minor surgery or having painful episodes without complications should not be transfused.
- Transfusion and splenectomy may be life-saving for young children with splenic sequestration. A full compatibility screen should always be performed.
- Folic acid 5 mg daily for life is recommended.
- Hydroxycarbamide (hydroxyurea)starting dose 20 mg/kg is the first drug which has been widely used as therapy for sickle cell anaemia. It acts by increasing Hb F concentrations but the reduction in neutrophils may also help. Hydroxycarbamide has been shown in trials to reduce the episodes of pain, the acute chest syndrome and the need for blood transfusions.
- Malaria prevention: Since they are more vulnerable to malaria, because the most common cause of painful crises in malaria countries is infection with malaria. It has therefore been recommended that people with sickle-cell disease living in malarial countries should receive anti-malarial chemoprophylaxis monthly for life i.e sulfadoxine pyrimethamine.
- Pain management.
- Home management
- Paracetamol 1 g every 8 hours
- Child: 10-15 mg/kg 6-8 hourly
- And/or ibuprofen Child: 5-10 mg/kg 8 hourly.
- Adults 400-600 mg 6-8 hourly.
- And/or diclofenac 50 mg 8 hourly
- Children only >9 years and >35 kg: 2 mg/kg in 3 divided doses.
- If pain not controlled, add:
- Codeine 30-60 mg every 6 hours (only in patients >12 years).
- Or tramadol 50-100 mg every 6-8 hours (only in patients >12 years)
- Or Oral morphine at 0.2-0.4 mg/kg every 4 hours and re-assess pain level.
If pain still not controlled, refer to hospital
At the hospital;
Morphine oral: Child and Adult: 0.3-0.6 mg/kg per dose and re-assess
Morphine Intravenously.
Child: 0.1-0.2 mg/kg per dose
Adult: 5-10 mg dose and re-assess
Use of laxative: bisacodyl 2.5 mg to 5 mg orally to prevent constipation due to morphine intake.
Cure:
- The only therapy approved by the FDA that may be able to cure SCD is a bone marrow or stem cell transplant.
- Bone marrow or stem cell transplants are very risky and can have serious side effects, including death. For the transplant to work, the bone marrow must be a close match. Usually, the best donor is a brother or sister.
Lifestyle Modifications:
- Regular Exercise: Moderate exercise, when tolerated, can improve cardiovascular health and reduce the risk of complications.
- Stress Management: Techniques like relaxation, meditation, and yoga can help manage stress levels and reduce the risk of crises.
- Healthy Diet: A nutritious diet rich in fruits, vegetables, and whole grains can support overall health.
- Avoidance of Extreme Temperatures: Extreme heat and cold can trigger crises.
- Altitude Management: Individuals should avoid high altitudes to minimize the risk of hypoxia.
Surgery:
- Bone Marrow Transplant: This is a potential cure, but it is a high-risk procedure with limited availability.
- Other Surgical Interventions: Surgical procedures may be necessary to correct bone deformities or treat complications like leg ulcers.
Support and Counseling:
- Genetic Counselling: Provides information about the inheritance of sickle cell disease and family planning options.
- Psychosocial Support: Provides emotional and practical support to help individuals cope with the challenges of living with sickle cell disease.
- Patient Education: Empowers individuals to manage their condition effectively by providing information on symptoms, triggers, and treatment options.
Prevention of Sickle cell crisis.
1. Hydration:
- Drink plenty of water: Staying well-hydrated is crucial for maintaining adequate blood flow and preventing sickling.
- Carry a water bottle and sip water regularly throughout the day.
- Avoid dehydration, especially during exercise, hot weather, or travel.
2. Temperature Management:
- Avoid extreme temperatures: Both excessive heat and cold can trigger sickle cell crises.
- Stay in air-conditioned environments during hot weather.
- Dress in layers to adjust to temperature changes.
- Be aware of the risk of hypothermia during cold weather.
3. Altitude Management:
- Avoid high altitudes: Low oxygen levels at high altitudes can worsen sickle cell symptoms.
4. Oxygen Management:
- Avoid situations with low oxygen levels: Avoid intense physical exertion, especially in hot, humid, or high-altitude environments.
- Use proper breathing techniques during exercise.
5. Infection Prevention:
- Vaccination: Receive all recommended vaccinations, including the pneumococcal vaccine, to protect against infections.
- Wash your hands frequently with soap and water.
- Use hand sanitizer when soap and water are unavailable.
- Avoid close contact with sick individuals.
- Practice safe food handling and preparation to prevent foodborne illness.
6. Routine Medical Care:
- Yearly visits to an eye doctor: Regular eye exams are crucial to monitor for signs of retinopathy, a serious complication of sickle cell disease.
- Regular checkups with a haematologist: Follow your doctor’s recommendations for regular blood tests and monitoring.
- Early intervention: Seek medical attention promptly for any unusual symptoms or signs of a sickle cell crisis.
7. Stress Management:
- Practice stress-reducing techniques: Stress can trigger sickle cell crises.
- Engage in activities you enjoy, like meditation, yoga, or spending time in nature.
- Seek counselling or therapy if you’re struggling to manage stress.
8. Lifestyle Modifications:
- Maintain a healthy weight: Obesity can worsen sickle cell symptoms.
- Eat a balanced diet rich in fruits, vegetables, and whole grains.
- Avoid smoking and excessive alcohol consumption.
- Get regular exercise, but consult your doctor about safe levels.
9. Advocacy and Support:
- Join a sickle cell support group: Connect with other individuals living with sickle cell disease and share experiences and resources.
Nursing Diagnosis
- Acute pain related to tissue hypoxia due to agglutination of sickled cells within blood vessels evidenced by patient verbalization.
- Risk for infection related to lowered immunity.
- Impaired Gas Exchange related to decreased oxygen-carrying capacity of the blood, reduced RBC life span/premature destruction, abnormal RBC structure; sensitivity to low oxygen tension (strenuous exercise, increase in altitude) as evidenced by difficulty in breathing.
- Ineffective Tissue Perfusion related to vaso-occlusive nature of sickling as evidenced by changes in vital signs: diminished peripheral pulses/capillary refill, general pallor or decreased mentation, restlessness.
- Risk for Deficient Fluid Volume related to increased fluid needs, e.g., hypermetabolic state/fever, inflammatory processes.
- Acute Pain related to Intravascular sickling with localized stasis, occlusion, and infarction/necrosis as evidenced by generalized pain, described as throbbing, or severe ; affecting peripheral extremities, bones, joints, back, abdomen, or head (headaches)
- Risk for Impaired Skin Integrity related to impaired circulation (venous stasis and vaso-occlusion)
Prevention Of Sickle Cell Disease
- Genetic counselling is important to prevent passing on the trait or disease to children for those wanting to have them.
- Premarital counselling is encouraged. Early recognition/screening of children with low Hb.
Complications of Sickle Cell anaemia
- Stroke. Issues in circulation will result to blockages, therefore predisposing the patient to develop thrombolytic strokes
- Acute chest syndrome. This is characterized by chest pain, fever and difficulty breathing requiring emergency medical treatment
- Pulmonary hypertension. This type of anaemia can cause build-up of unnecessary lung pressure due to problems with circulation as a result of erythrocyte clumping
- Organ damage. Due to the chronic inability of the red blood cells to provide essential oxygen for normal organ function, patients with sickle cell anaemia may develop organ failure, which can be fatal.
- Blindness. One of the potential complications of having abnormal red blood cells circulating in the body is damage to smaller blood vessels, particularly the eye. This in turn will cause eye damage and eventually blindness.
- Leg ulcers. Poor wound healing and rampant skin breakdown can be observed for patients suffering from sickle cell anaemia.
- Gallstones. The build of bilirubin caused by the metabolism of the abnormal erythrocytes will result to gall stones that will block the flow of bile.
- Priapism. This is a condition wherein men with Sickle cell anaemia will present with painful and long-lasting erections due to the blockages of the tiny blood vessels of the penis.
- Pregnancy complications. Sickle cell anaemia increases the risk of high blood pressure and the presence of clots that will interfere with the normal development of the fetus.
NURSING CARE PLAN FOR A PATIENT WITH SICKLE CELL CRISIS
|
Assessment |
Diagnosis |
Goals/Expected Outcomes |
Intervention |
Rationale |
Evaluation |
|
Cyanosis, breathlessness at a rate of 28 breaths/min, restlessness, and SpO2 of 80%. |
Impaired gaseous exchange related to increased viscosity of blood evidenced by cyanosis, breathlessness, restlessness, and SpO2 of 80%. |
– Establish adequate gaseous exchange within 2 hours. – Improve SpO2 by 10% within the first 30 minutes. – Establish a normal breathing pattern without assisted respiration within 1 hour. – Restore normal skin color in 30 minutes. |
– Establish an intravenous line and administer fluids (normal saline 500 mL every 6 hours for 24 hours). – Encourage fluid intake by mouth. – Start a fluid input and output chart. – Assess the need for more fluids after 24 hours. – Take vital signs every 30 minutes for 2 hours, paying attention to breathing and SpO2, then adjust according to findings. – Administer oxygen 3 L/min for 1 hour using a face mask. |
– Establishing IV access and administering fluids help to reduce blood viscosity and improve circulation. – Encouraging oral fluid intake promotes hydration. – Fluid balance chart helps to monitor fluid status. – Regular assessment ensures timely adjustments in fluid therapy. – Oxygen therapy increases oxygen saturation in the blood. |
– Patient is resting. – Normal breathing pattern restored, rate 20 breaths/min. – SpO2 improved to 98% on room air. – Normal skin colour restored, lips look pink. |
|
Patient verbalizing throbbing pain in the legs and joints, rating score of 8 on the pain scale. |
Acute pain related to intravascular sickling with localized stasis evidenced by patient verbalizing throbbing pain in the legs and joints. |
– Relieve pain within 4 hours. – Improve venous patency – Improve circulatory flow. |
– Administer analgesia (pethidine 50 mg single dose, then tramadol 50 mg every 8 hours for 3 days as prescribed and document). – Continue intravenous fluids as above and monitor pain hourly. |
– Analgesics provide comfort and relieve restlessness. – IV fluids maintain normal circulatory flow. |
– Patient reports pain relief after 4 hours, score 2 on the pain scale. |
|
Reduced haemoglobin levels of 5 g/L according to laboratory results, swelling of the lower limbs and joints. |
Altered tissue perfusion related to decreased red blood cells as evidenced by reduced haemoglobin levels of 5 g/L, swelling of the lower limbs and joints. |
– Restore normal tissue perfusion within 24 hours. – Establish normal tissue perfusion. |
– Transfuse with units of packed cells 5 mL/kg/h as prescribed. – Continue with fluid balance chart. – Apply a warm compress to the affected areas. – Elevate the affected limbs. |
– Blood transfusion increases haemoglobin levels. – Fluid balance chart monitors fluid status. – Warm compresses promote vasodilation and circulation to hypoxic areas. – Elevation reduces swelling and promotes venous return. |
– Increased haemoglobin levels of 7 g/dL as seen in post-transfusion lab report. – Swelling has subsided, and the patient is able to move the limb. |
|
Fever, hypermetabolic state, dehydration symptoms (dry mucous membranes, poor skin turgor). |
Risk for fluid volume deficit related to increased fluid needs due to hypermetabolic state or fever. |
– Maintain adequate hydration. – Prevent fluid volume deficit. |
– Monitor vital signs and fluid status regularly. – Encourage oral fluid intake and administer IV fluids as needed. – Educate the patient on the importance of fluid intake. |
– Regular monitoring detects early signs of fluid deficit. – Ensuring adequate hydration prevents complications. |
– Fluid balance is maintained, and signs of dehydration are absent. |
|
Presence of venous stasis, vaso-occlusion, decreased mobility, and risk of skin breakdown. |
Risk for impaired skin integrity related to impaired circulation due to venous stasis and vaso-occlusion, and decreased mobility. |
– Prevent skin breakdown. – Maintain skin integrity. |
– Assess skin regularly for signs of breakdown. – Reposition the patient every 2 hours. – Provide skin care and keep the skin clean and dry. – Use pressure-relieving devices as needed. |
– Regular assessment and repositioning prevent pressure ulcers. – Good skin care promotes skin integrity. |
– Skin remains intact without signs of breakdown. |
Quick Quiz
Sicklecell Disease Quiz
Paediatrics - mobile-friendly and focused practice.
Privacy: Your details are used only for quiz tracking and certificates.
Sicklecell Disease Quiz
Paediatrics
Preparing questions...
Choose your answer and keep your streak alive.
Great effort.
Here is your quick performance summary.
It’s an interesting and enjoyable platform thanks alot
Thanks for the great w
Thanks, interesting revision
Well explained established notes
it’s really good for revision
thanks
Welcome, anything you wish we can improve?
Thanks for the knowledge
Thank you so much for the effort
May the Almighty God bless you
Really updated revision, thanks so much
I love it every tym I open these notes
So enjoyable and helpful for me currently doing my extension in nursing diploma
Thanks nice presentation
𝐭𝐡𝐚𝐧𝐤𝐬 𝐚𝐥𝐨𝐭
Thank you.
Thanks for the great work.may the almighty God bless you abundantly 😊🙏
thanx for the knowladge obtained;
GOD BLESS YOU
AMEN
Nursing diagnosis is still my problem
Try with your tutor to understand the objective and subjective data, and understand the question, it’s really going to be smooth for you dear
I highly appreciate, you make learning a smooth journey for most of us .
But include other notes too ,, otherwise these notes are the best