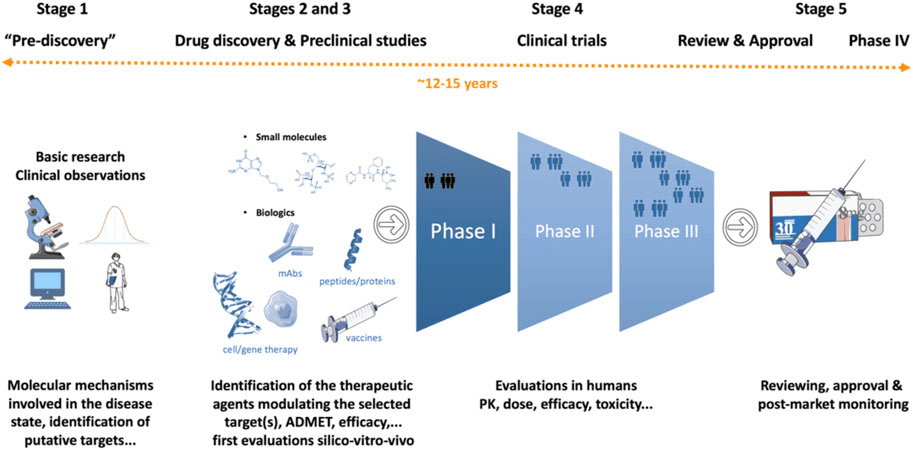
The Drug Development Process
The Drug Development Process
The Drug Development Process
Bringing a new drug to the pharmacy shelf is not a simple laboratory experiment; it is a massive, highly regulated journey. This guide will break down the entire process from a simple idea in a lab to post-marketing surveillance. Examiners love to test your knowledge on the differences between the Clinical Trial Phases (I, II, III, and IV), the definition of a new drug, and the "Pyramid of Uncertainty." Pay close attention to the scenarios provided, as they will help you remember the dry facts.
1. Introduction: The Pyramid of Uncertainty
The development of a new drug is an incredibly time-consuming and extremely expensive process. During the last 50 years, hundreds of new drugs have been introduced to save lives, while many older drugs have been entirely deleted (withdrawn) from the market due to newly discovered toxicities or better alternatives.
We call this the "Pyramid of Uncertainty" because the failure rate is exceptionally high. Less than 1% (<1%) of compounds that go into testing eventually become licensed, usable medicines.
The Timeline and Attrition Rate
To successfully bring just one single new drug to the market, it requires a deep understanding of both the development process and the integral role that preclinical (animal/lab) testing plays. Let's look at the numbers:
- Time: It takes 10 to 12 years (sometimes up to 24 years from the initial idea) on average for an experimental drug to travel from the laboratory bench to the patient's medicine cabinet.
- Success Rate: Out of 5,000 to 10,000 compounds screened during initial discovery, only about 250 will make it to preclinical (animal) testing.
- From those 250, only FIVE (5) compounds will be deemed safe enough to enter human clinical trials (Phase I).
- Out of those 5 compounds tested in humans, only ONE (1) is finally approved by regulatory bodies.
| Stage of Development | Number of Compounds Surviving | Failure Rate at this Stage |
|---|---|---|
| Discovery / Idea | 5,000 - 10,000 | N/A |
| Preclinical Testing | 250 | 50% fail here |
| Phase I (Clinical) | 5 | 30% fail here |
| Phase II (Clinical) | 1 (sometimes 2) | 50% fail here |
| Phase III (Clinical) | 1 | - |
| FDA Review & Approval | 1 Product Licensed | - |
2. What is a "New Drug" and Who Regulates It?
Definition of a NEW DRUG
In pharmacology and law, a "new drug" does not just mean a chemical that was invented yesterday. The legal definition encompasses three specific scenarios:
- A Completely New Substance: A chemical entity which, except during local clinical trials, has never been used before in the country.
- An Already Approved Drug with NEW CLAIMS: If a drug is already on the market, but the manufacturer wants to market it with modified or new claims. This includes a new indication (what disease it treats), a new dosage, a new dosage form (changing from a tablet to an IV injection), or a new route of administration.
Minoxidil was originally approved as an oral tablet to treat high blood pressure. Later, researchers discovered it caused hair growth. When the company wanted to sell it as a topical lotion for baldness (new indication, new route, new dosage form), it had to go through the approval process again as a legally "New Drug."
- Fixed-Dose Combination (FDC): Two or more already known and approved drugs proposed to be combined for the very first time in a single pill at a fixed ratio.
Drug A (Artemether) and Drug B (Lumefantrine) are both known malaria drugs. If a company decides to combine them into one single tablet (Coartem), that combination is legally considered a "New Drug" and must be tested to ensure the two chemicals don't react toxically with each other inside the pill.
Regulatory Authorities and Guidelines
Every country has a strict police force for medicines to protect the public. They issue guidelines on clinical trials that are required to be carried out before a drug can be imported or manufactured.
- Uganda: NDA (National Drug Authority). The power to grant permission for a new drug to be tested and marketed in Uganda rests solely with the NDA, governed by the NDA Act. The NDA Act details exactly what preclinical (animal) data is required before human tests begin.
- United States: US-FDA (Food and Drug Administration).
- Europe: EMEA (European Medicines Agency).
- United Kingdom: MHRA (Medicines and Healthcare products Regulatory Agency).
- Japan: MHLW (Ministry of Health, Labour and Welfare).
- Australia: TGA (Therapeutic Goods Administration).
3. The 8 Steps in New Drug Development
The journey follows a strict chronological order:
- Idea or Basic Research
- New Drug Discovery
- Screening
- Preclinical Studies
- Formulation Development
- IND (Investigational New Drug) Application
- Clinical Studies (Human Trials)
- Official License / Regulations / Marketing
4. Step A & B: Basic Research and New Drug Discovery
A. Basic Research
Before you can invent a drug, you must thoroughly understand the disease.
- Start by studying normal and abnormal body functions.
- Investigate each component of the disease (its pathophysiology). Ask questions: What are the symptoms? What is the root cause? Which is the target organ? What are the biochemical pathways involved?
- Look up information obtained in previous research and publications.
- Find out at exactly which stage we can stop the disease progression. This becomes OUR TARGET!
- Search for a targeted drug, isolate the index compound, perform early animal testing for safety, and eventually seek approval to test in humans.
B. New Drug Discovery (4 Sub-steps)
Once the research is done, the actual discovery phase begins, taking roughly 5 to 6 years.
- Target Identification: Choosing a specific cellular or genetic chemical within our body (the "target") that is associated with the disease.
- Target Validation: Checking and confirming that interacting with this specific target actually changes the disease condition. (Analogy: Making sure you have found the correct lock before you start building keys.)
- Lead Identification: Finding a "Lead compound." A lead is a substance believed to have the potential to treat the disease. Scientists use massive collections (libraries) of up to 5,000-10,000 molecules. Each molecule is rigorously tested to confirm its effect on the target.
- Lead Optimization: Comparing the properties of various successful lead compounds. This provides information to help pharmaceutical companies select the single compound with the greatest potential to become a safe, effective medicine. During this stage, Lead Prioritization Studies are conducted in living organisms (in vivo) and in test tubes/cells (in vitro) to compare their metabolism and effects.
Characteristics of an Ideal Drug Candidate
During lead optimization, scientists are looking for a molecule that possesses these "perfect" traits:
- High Potency: Only a small amount is needed to produce the desired effect.
- High Selectivity: It attacks ONLY the disease target and leaves normal, healthy cells alone.
- Good Oral Bioavailability: It can be swallowed as a pill and successfully reach the bloodstream, rather than needing to be injected.
- Low or no interaction with CYP450: CYP450 are liver enzymes. If a drug interacts heavily with them, it will cause severe drug-drug interactions with other medicines the patient is taking.
- Less or minimal adverse (side) effects.
- Good Therapeutic Index: There is a very large, safe gap between the dose that cures the patient and the dose that poisons the patient.
5. Step C & D: Screening and Pre-clinical Studies
C. Screening
New Chemical Entities (NCEs) are subjected to a battery of rapid screening tests to quickly identify active compounds, antibodies, or genes which modulate a biological pathway. This is done on animal behavior, isolated tissues, and intact animals. Remember: 1 in every 4,000-5,000 NCEs screened is actually marketed.
D. Pre-clinical Studies (Animal Testing)
Before a drug is allowed anywhere near a human, it must be tested heavily in the lab. This takes years. Tests are conducted on:
- Isolated organs.
- Bacterial cultures (to check for genetic mutations/cancer-causing potential).
- Intact animals (general observational tests).
- Animal Models of Human Diseases: Scientists artificially induce human diseases in animals to see if the drug cures them.
- Exam Example 1: Using diazoxide to induce diabetes in rats/dogs, then testing a new anti-diabetic drug on them.
- Exam Example 2: Using "kindled animals" (animals whose brains have been stimulated to have seizures) to test new anti-epileptic drugs.
Pre-clinical testing is divided into two main categories:
- Pharmacologic Studies: Looking at what the drug does and how it does it. Evaluates specific biological activities, mechanism of action, Pharmacokinetics (PK - absorption, distribution, metabolism, excretion), and effective dose range. At the cellular level, scientists determine if the drug acts as a Receptor agonist/antagonist (checking affinity and selectivity) or as an Inhibitor of a key enzyme.
- Toxicity Studies: Looking at how dangerous the drug is. Evaluates potential risks. The goals are identifying safe drugs, identifying potential human toxicity, and predicting specific toxicities to be closely monitored when clinical trials finally begin.
6. Step E & F: Formulation Development and IND Application
E. Formulation Development
People do not swallow pure chemical powder; they take a formulated medicine. Formulation is mixing the pure DRUG + Additives (Excipients).
- Additives include: Fillers (to add bulk to tiny drug amounts), lubricants (so pills don't stick to factory machines), coatings, stabilisers (so it doesn't expire quickly), colours, binders (to hold the pill together), and disintegrators (to make the pill explode and dissolve once it hits stomach acid).
- Dosage Form: Deciding if it will be a capsule, tablet, or injection.
- Manipulation: Designing the formulation to manipulate the drug's profile, such as creating a sustained release tablet, ensuring good bioequivalence, bioavailability, and ease of use for the patient.
F. Investigational New Drug (IND) Application
After successful preclinical (animal) development, the sponsor (pharmaceutical company) compiles all their data into an IND application.
What is an IND?
An IND is a vehicle through which the sponsor asks permission to advance to the next stage: clinical (human) trials. It is NOT an application for marketing or selling the drug. It is simply asking: "We have tested this on rats and it looks safe, please let us test it on humans."
- Contents of an IND: Animal pharmacological & toxicology studies, manufacturing information, clinical protocols (how they plan to test humans safely), and investigator information.
- Sponsor/FDA Pre-IND Meeting: Prior to clinical studies, the sponsor needs evidence the compound is biologically active and reasonably safe for initial human administration. Meeting at this early stage provides an open discussion about testing phases, data requirements, and resolving scientific issues prior to the formal IND submission.
7. Step G: Clinical Studies (Phases I, II, and III)
Once the IND is approved by the regulatory authority and ethics committees, Human Clinical Pharmacology Studies begin. These take 3 to 10 years.
The "Is it safe?" Phase
- Participants: Conducted on a small group of 20 to 80 HEALTHY human volunteers.
- Exception: For highly toxic drugs, like anti-cancer/chemotherapy drugs, Phase I is conducted on sick patients, because it is unethical to give highly toxic poison to a healthy person.
- Place: Special testing facilities where participants are monitored exceptionally closely by physicians and trained investigators.
- Objectives:
- Determine Safety and Tolerability.
- Find the Maximum Tolerated Dose (MTD) before side effects become unacceptable.
- Determine Pharmacokinetics (PK) and Pharmacodynamics (PD) in the human body.
- Measurement of drug activity.
The "Does it work?" Phase
Known as Therapeutic Exploratory Trials.
- Participants: This is the first trial in PATIENTS actually suffering from the disease to be treated. Uses 50 to 300 patients.
- Place: Specialized hospital units with closely monitored physicians and trained investigators.
- Objectives:
- Determine the Effectiveness of the drug (Proof of Concept).
- Identify common short-term side effects and risks.
- Determine exact therapeutic regimens, plans, and doses required for the upcoming Phase III trials.
- Additional PK, PD, and safety evaluations.
- Identify the specific target populations for further studies.
- End of Phase 2 Meeting (Sponsor/FDA): One month prior to the end of Phase 2, the sponsor submits background info and protocols for Phase 3. This allows the review team to prepare for a productive meeting before launching massive Phase 3 trials.
The "Is it better?" Phase
Initiated only when Phase II data shows solid evidence of efficacy.
- Participants: A massive group of 300 to 3,000+ patients (sometimes up to 10,000).
- Place: These are Multi-centric (conducted at multiple hospitals simultaneously across the country or world). Because of multiple sites and huge numbers, this is the most expensive and most time-consuming phase.
- Design: Prospective, Randomized, Controlled trials.
What does this mean? A large sample population is randomly split. Group A (Intervention) gets the new drug. Group B (Control) gets a Placebo or the current standard medication. We measure the % who get better to prove it wasn't just the placebo effect. - Types of Phase III:
- Phase IIIa: Carried out on a large number of patients. Strict regulatory requirement needed to submit the New Drug Application (NDA).
- Phase IIIb: Extended trials conducted after applying for approval, but before launch.
- Objectives:
- Confirm efficacy and safety profile on a large population.
- Comparison with the current Gold Standard treatment.
- Identify specific disease sub-types for which the drug is most effective.
- Provide hard data to write the product package insert.
8. Step H: Official License / Marketing (NDA) & Phase IV
The New Drug Application (NDA)
Once Phase III is complete, the sponsor submits the NDA.
- The NDA is a massive vehicle/document through which sponsors formally propose that the FDA approve the new pharmaceutical for public sale.
- It documents safety and efficacy and contains all the information collected during the entire 10-12 year Drug Development Process.
- The FDA Review Process for granting marketing permission takes between 0.5 to 2 years (six months to two years).
Phase IV (Post-Marketing Surveillance - PMS)
Just because the drug is in the pharmacy does not mean the testing is over. Phase IV constitutes vigilant Post-Marketing Surveillance (PMS) to continuously monitor the safety of the new drug in the real world.
- Participants: Patients receiving the marketed drug for actual therapy (2,000 to 10,000+).
- Players: Principal investigators, General Practitioners (regular doctors), and specialists.
- What is PMS? A systemic method for the continuous surveillance of adverse reactions, observing patterns of drug utilization, and discovering additional indications.
- Objectives:
- Confirm efficacy and safety in massive, diverse populations during real-world medical practice.
- Detect rare, unknown adverse drug reactions. (Example: A side effect that only happens 1 in 50,000 times will never be seen in a Phase III trial of 3,000 people. It will only be caught in Phase IV when millions take the drug.)
- Evaluate what happens during over-dosage and when taken concomitantly (at the same time) with other treatments.
- Identify new indications (new diseases the drug might cure).
- Evaluate Pharmacoeconomics (is the drug cost-effective for society?).
9. Timelines of Patent & Modern Advances
Timelines of Patent
Inventing a drug costs billions of dollars. To allow companies to recover their money, governments grant them a Patent for 20 years. During this time, the pharmaceutical company has exclusive rights to produce and sell the drug. No one else can copy it.
Because the 20-year patent clock starts ticking during the early discovery phase, by the time the drug finishes the 12-year clinical trial process and gets approved, the company may only have 8 years left of exclusive sales. After the expiry of the patent, any other company may legally produce and market the exact same drug as a cheap generic product (like generic paracetamol or amoxicillin).
Advances in Drug Development: Phase 0 Microdosing
Because clinical trials are so expensive and the failure rate is so high, scientists have developed a new, highly advanced step called Phase 0 Microdosing.
- What is it? A new approach to obtain human pharmacokinetic (PK) information before the usual, highly expensive Phase I safety program is conducted.
- The Dose: It uses minute, tiny quantities of the drug—specifically 1/100th of the dose that is anticipated to produce a pharmacological effect.
- The Goal: Because the dose is so small, it is not intended to produce any pharmacologic effect (no curing), and therefore, it will not cause any adverse side effects. However, modern sensitive instruments can trace this microdose to provide incredibly useful Pharmacokinetic info.
- Why do it? It is hypothesized that microdosing will help reduce or entirely replace the extensive, costly animal testing needed for kinetics. It provides enough data to decide if the drug is worth pushing forward.
- The Benefit: It helps in early de-selection. If the human body immediately destroys the microdose, the company drops the drug immediately. This creates massive cost savings related to manufacturing, scaling up, and running full trials for a drug that was destined to fail anyway.
Quick Quiz
Palliative Care Day 7 Quiz
Palliative Care - mobile-friendly and focused practice.
Privacy: Your details are used only for quiz tracking and certificates.
Palliative Care Day 7 Quiz
Palliative Care
Preparing questions...
Choose your answer and keep your streak alive.
Great effort.
Here is your quick performance summary.
The Drug Development Process Read More »