Introduction to Disaster in Nursing
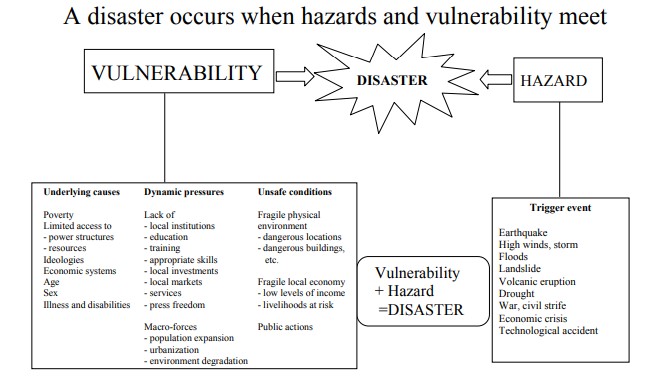
A disaster is an occurrence that disrupts the normal conditions of existence and causes a level of suffering that exceeds the capacity of adjustment of the affected community.
A disaster is a sudden or unexpected catastrophic event that causes serious disruption of the functioning of a community or society. The disruption is so severe that it exceeds the ability of the affected community to cope using its own resources.
| Element | Simple Explanation | Ugandan Example |
|---|---|---|
| Disruption of normal life | Daily activities stop or become very difficult | When Bududa landslides occur, people cannot farm, children cannot go to school, and markets close |
| Exceeds community capacity | The community cannot handle it alone | A village clinic in Kasese being overwhelmed by hundreds of flood victims at once |
| Requires external help | Help must come from outside | When Ebola struck Uganda, teams from Kampala, WHO, and MSF had to come to help local health workers |
| Causes suffering | People experience death, injury, or loss | Families in Mbale losing homes, crops, and loved ones during flash floods |
"If the community can cope, it is an EMERGENCY. If the community CANNOT cope, it is a DISASTER."
Vulnerability is the lack of capacity to deal with a potential threat. It means not having enough information, resources, or technology to protect yourself from harm.
Think of vulnerability as being unprotected or exposed to danger. A person or community is vulnerable when they do not have what they need to stay safe during a disaster.
Vulnerability comes from many sources:
- Physical factors: Where you live, how your house is built
- Social factors: Your relationships, education, and community support
- Economic factors: How much money and resources you have
- Environmental factors: The condition of the land, water, and air around you
- Poor design and construction of buildings (mud-and-wattle houses on steep hills)
- Lack of public information and awareness (communities not knowing landslide warning signs)
- Limited official recognition of risks (no early warning systems in place)
- Disregard for environmental management (cutting down trees on hillsides, leading to soil erosion)
Definition: Physical vulnerability refers to how the physical environment and infrastructure put people at risk. It is determined by population density, how remote an area is, the site of buildings, and the materials used for construction.
Factors Affecting Physical Vulnerability:
- Population density: More people living closely together means more people can be hurt at once
- Remoteness: Areas far from hospitals and roads are more vulnerable because help cannot arrive quickly
- Building design: Houses built without proper foundations or support
- Construction materials: Weak materials that cannot withstand disasters
- Infrastructure quality: Poor roads, weak bridges, unreliable communication systems
| Situation | Why It is Physically Vulnerable | What Can Happen |
|---|---|---|
| Wooden homes | Wood is light and flexible but burns easily | In an earthquake, wooden homes may not collapse, but if there is a fire afterward, they burn quickly |
| Mud-and-wattle houses on hillsides | Mud dissolves in heavy rain; steep slopes are unstable | During rains in Bududa, these houses slide down with the soil |
| Houses built in wetland areas | Wetlands flood easily; foundations become weak | In Kampala's Bwaise area, homes flood every rainy season |
| Remote villages in Karamoja | Far from hospitals, no ambulances, poor roads | During drought, malnourished children cannot reach health centers in time |
Definition: Social vulnerability is the inability of people, organizations, and societies to withstand adverse impacts from hazards because of characteristics in their social interactions, institutions, and cultural values.
What Creates Social Vulnerability? Social vulnerability is about people and how they live together. It includes:
- Levels of literacy and education: People who cannot read may not understand warning signs or evacuation instructions. Educated communities are more likely to prepare for disasters.
- Peace and security: Communities experiencing conflict are more vulnerable because they are already stressed. Displacement camps are socially vulnerable environments.
- Access to basic human rights: If people do not have rights to land, housing, or healthcare, they cannot protect themselves. Marginalized groups often live in unsafe areas because they have no choice.
- Good governance: Communities with strong local leaders and clear disaster plans are less vulnerable. Corruption and poor planning increase vulnerability.
- Social equity: When some groups are treated unfairly, they become more vulnerable. Gender inequality can make women and girls more vulnerable during disasters.
- Traditional values and beliefs: Some beliefs may prevent people from evacuating or seeking medical help. Positive traditions (like community cooperation) can reduce vulnerability.
| Group | Why They Are Socially Vulnerable | Real-Life Scenario |
|---|---|---|
| Children | Cannot make decisions for themselves; depend on adults | During flooding in Teso, children may be left home alone while parents search for food |
| Elderly people | May be unable to walk quickly or understand warnings | An old woman in Kisoro may not hear the landslide warning if she lives alone and has no radio |
| People with disabilities | May not be able to evacuate without assistance | A blind person in a Kampala slum cannot find the evacuation route during a fire |
| Pregnant women | Slower movement, special medical needs | A pregnant mother in a remote village cannot run from floodwaters and needs antenatal care |
| Refugees and internally displaced persons | No permanent home, limited social networks | South Sudanese refugees in Ugandan camps are vulnerable to disease outbreaks |
Definition: Economic vulnerability means that the level of risk depends heavily on how much money people, communities, or nations have. Poor people are usually more vulnerable to disasters because they lack resources to protect themselves.
Why Poverty Creates Vulnerability:
- Cannot afford safe housing: Poor families build houses with cheap materials in dangerous locations. They cannot afford to reinforce buildings against earthquakes or floods.
- Cannot afford insurance: When disaster strikes, they lose everything with no way to recover. No savings to rebuild homes or replace lost crops.
- Depend on daily wages: If work stops because of disaster, they immediately have no food. Cannot stockpile food or medicine for emergencies.
- Limited access to healthcare: Cannot pay for transportation to hospitals. Cannot afford medicines or treatments after injury.
| Economic Situation | Vulnerability | Ugandan Example |
|---|---|---|
| Poor family | Lives in unsafe area because it is cheap | A family in Kampala's Bwaise lives in a wetland because they cannot afford rent in safer areas like Ntinda |
| Wealthy family | Can afford safe housing and insurance | A family in Kololo has a strong concrete house on high ground with emergency supplies |
| Poor nation | Limited resources for disaster preparedness | Uganda has fewer emergency helicopters and advanced rescue equipment compared to developed countries |
| Subsistence farmer | Loses entire livelihood when crops are destroyed | A farmer in Gulu who loses their maize crop to drought has no food and no income for the year |
↓
POOR PEOPLE LOSE HOMES, CROPS, JOBS
↓
THEY BECOME EVEN POORER
↓
THEY MOVE TO EVEN MORE DANGEROUS AREAS
↓
THEY BECOME MORE VULNERABLE TO THE NEXT DISASTER
↓
(REPEAT)
Definition: Environmental vulnerability refers to the depletion and degradation of natural resources that would otherwise protect communities from disasters. When the environment is damaged, disasters become worse.
Key Aspects of Environmental Vulnerability:
- Natural resource depletion: Cutting down forests (deforestation), draining wetlands, overusing groundwater.
- Resource degradation: Soil erosion from poor farming practices, water pollution from industrial waste.
- Loss of biodiversity.
| Environmental Damage | How It Increases Disaster Risk | Ugandan Example |
|---|---|---|
| Deforestation on hillsides | Tree roots hold soil together; without trees, soil slides in rain | Mt. Elgon slopes in Bududa were deforested, leading to deadly landslides |
| Wetland destruction | Wetlands absorb floodwater; without them, floods are worse | Kampala's Nakivubo and Lubigi wetlands were drained, causing worse flooding in Bwaise and Kalerwe |
| Overgrazing | Grass holds topsoil; without it, soil blows away or washes away | In Karamoja, overgrazing has led to desertification and worse drought impacts |
| Water pollution | Contaminated water spreads disease after disasters | After floods in Mbale, polluted water caused cholera outbreaks |
| Climate change | Changes weather patterns, making extremes more common | Increasingly unpredictable rainy seasons in Uganda |
The Caroni Swamp Example (International Context): Wetlands like the Caroni Swamp are sensitive to increasing salinity from seawater, pollution from stormwater runoff containing agricultural chemicals, and eroded soils flowing into the wetland. This shows how environmental damage in one area affects another.
Definition: Disaster risk is the likelihood that a specific hazard will occur in a particular place and the probable consequences for people, property, and the environment.
Simple Explanation: Risk = How likely something bad is to happen + How bad it will be if it happens.
Components of Disaster Risk:
- Hazard: The dangerous event itself (e.g., earthquake, flood)
- Exposure: People and property in the hazard's path
- Vulnerability: How susceptible those people and properties are
- Capacity: The ability to cope and recover
Definition: Acceptable risk is the level of risk that a community is willing to tolerate given their social, economic, political, cultural, technical, and environmental conditions.
Simple Explanation: Some risks are so small or so much a part of daily life that people accept them.
| Activity | Risk | Why It is Acceptable |
|---|---|---|
| Flying in an airplane | Crash | Very rare; benefits of travel outweigh the small risk |
| Eating street food | Food poisoning | Small risk; food is affordable and convenient |
| Living in a mild earthquake zone | Minor tremors | Earthquakes are rare and usually weak |
| Crossing a busy road in Kampala | Accident | Necessary for daily life; people accept the risk |
Important Note: What is acceptable in one community may not be acceptable in another. Wealthy communities may demand zero risk, while poor communities may accept higher risks because they have no alternatives.
Definition: Residual risk is the disaster risk that remains even after effective disaster risk reduction measures have been put in place. This is the risk for which emergency response and recovery capacities must always be maintained.
Simple Explanation: Even when you do everything right, some danger always remains. You cannot eliminate all risk.
| Mitigation Measure | Residual Risk | Explanation |
|---|---|---|
| Building codes for earthquakes | House still destroyed by massive quake | Codes help with moderate quakes, but not the strongest possible ones |
| Drainage systems in Kampala | Flooding still occurs in extreme rainfall | Drains handle normal rain, but not record-breaking storms |
| Vaccination programs | Disease outbreak still possible | Vaccines are not 100% effective; new strains may emerge |
| Early warning systems for landslides | Some people still do not evacuate in time | Warnings may come too late, or people may not believe them |
Definition: Intensity refers to a disaster agent's ability to inflict damage and injury. It measures how strong or severe the disaster is.
| Disaster | Low Intensity | High Intensity |
|---|---|---|
| Earthquake | Minor tremor; cracks in walls | Major quake; buildings collapse |
| Flood | Water reaches ankles | Water reaches rooftops |
| Windstorm | Branches break | Trees uprooted, houses destroyed |
| Disease outbreak | Few cases, mild symptoms | Many cases, severe symptoms, deaths |
Definition: Scope refers to the geographic area and social space impacted by the disaster agent. It answers the question: "How wide an area is affected?"
- Narrow scope: Affects one building, one street, or one village
- Moderate scope: Affects a district or region
- Broad scope: Affects multiple regions or the entire country
| Disaster | Scope | Description |
|---|---|---|
| House fire in Jinja | Narrow | One family affected |
| Bududa landslide | Moderate | Several villages in one district |
| COVID-19 pandemic | Broad | Entire country and world affected |
| Drought in Karamoja | Moderate to Broad | Entire region, multiple districts |
Definition: Frequency refers to the number of times certain disasters occur in specific geographical locations. It tells us how often a disaster happens in the same place.
- High frequency gives communities experience and may lead to better preparation.
- Low frequency means communities may forget past disasters and become complacent.
- Frequency data helps planners decide where to invest in prevention.
| Location | Disaster | Frequency | Community Response |
|---|---|---|---|
| Bududa, Mbale | Landslides | Almost every rainy season | Some preparedness; early warning systems being developed |
| Karamoja | Drought | Regular, cyclical | Communities have some coping strategies but remain vulnerable |
| Kampala (Bwaise) | Flooding | Every heavy rainfall | Known risk, but poverty keeps people living there |
| Rwenzori region | Earthquakes | Rare | Low awareness and preparation |
Definition: Controllability refers to the control measures that can reduce the impact of a disaster. It helps emergency planners know what actions will be effective.
| Level | Description | Example |
|---|---|---|
| Highly controllable | Human actions can prevent or greatly reduce impact | Industrial accidents with safety protocols; disease outbreaks with vaccination |
| Moderately controllable | Some impact can be reduced, but not all | Flooding with drainage systems; earthquakes with building codes |
| Not controllable | Little or nothing can be done to stop it | Major earthquakes; volcanic eruptions; hurricanes |
Definition: Triage is the sorting of victims according to the extent of severity of their injuries or conditions. It helps decide who gets treated first when resources are limited.
Why Triage is Critical: In a disaster, there are often too many patients, too few medical staff, too little equipment, and too little time. Triage ensures that the greatest number of lives are saved with available resources.
| Color | Category | Description | Priority | Example |
|---|---|---|---|---|
| 🔴 RED | Immediate | Life-threatening but treatable | FIRST | Severe bleeding, airway obstruction, shock |
| 🟡 YELLOW | Delayed | Serious but stable for now | SECOND | Broken bones, deep wounds without active bleeding |
| 🟢 GREEN | Minor | Walking wounded; can wait | THIRD | Minor cuts, bruises, anxiety |
| ⚫ BLACK | Deceased/Expectant | Dead or dying; resources would be wasted | LAST (or none) | No pulse, no breathing, severe burns over 90% body |
- Rapid assessment – 30-60 seconds per patient
- Check airway, breathing, circulation (ABC)
- Assign color tag
- Move to appropriate area
- Re-triage regularly – Conditions change
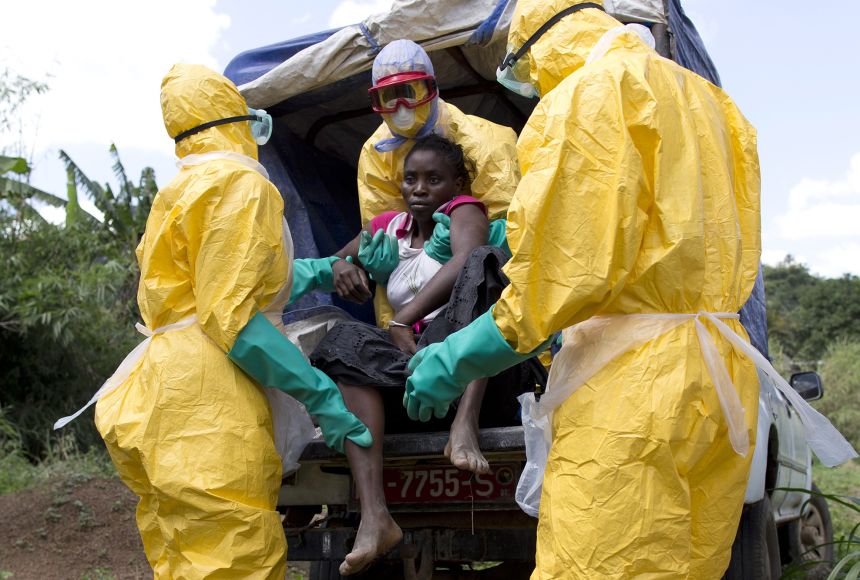
Ugandan Context Example: During the 2010 Kampala bombings, nurses and doctors had to triage victims at Mulago Hospital. Those with severe bleeding (RED) were taken to surgery first. Those with minor injuries (GREEN) waited and helped comfort others.
Definition: Time refers to the period when certain disasters can last and the warning period that allows people to evacuate or prepare.
Types of Time Factors: Warning time (advance notice), Duration (how long it lasts), Speed of onset (how quickly it happens).
| Disaster | Warning Time | Duration | Speed of Onset |
|---|---|---|---|
| Hurricane/Cyclone | Hours to days | Hours to days | Slow |
| Flood | Hours to days | Days to weeks | Moderate |
| Landslide | Minutes to hours | Minutes | Fast |
| Earthquake | Seconds to none | Seconds to minutes | Very fast |
| Drought | Months | Months to years | Very slow |
| Disease outbreak | Days to weeks | Weeks to months | Moderate |
Definition: Capacity is the ability of a community to use all available resources to reduce risk levels and disaster effects.
| Type | Example in Uganda |
|---|---|
| Physical capacity | A district hospital with a generator, water tank, and emergency stockpile |
| Human capacity | Community health workers trained in first aid |
| Organizational capacity | The Office of the Prime Minister's disaster preparedness unit |
| Social capacity | Village savings groups that can quickly lend money after a disaster |
Definition: Capacity building is the efforts to develop human skills within a community to reduce risk levels. It is about making people and communities stronger and better prepared.
- Methods: Training (first aid, search and rescue), Education (school programs), Drills and simulations, Resource provision, Institutional strengthening.
Definition (WHO): An emergency is a state in which normal procedures are suspended and extraordinary measures are taken in order to avert a disaster.
Simple Explanation: An emergency is a serious situation that requires immediate action, but the community CAN still handle it with its own resources.
- Predictable and narrow in scope
- Standard procedures are sufficient
- Local resources can manage it
- Examples: house fire, vehicle accident, single building collapse
Definition: A catastrophe is a large-scope event that affects multiple communities, produces very high levels of damage and social disruption, and sharply and concurrently interrupts community and lifeline services.
- Multiple communities affected – Not just one village or district
- Very high damage – Destruction on a massive scale
- Lifeline services interrupted – No water, electricity, communication, transportation
- Emergency response systems overwhelmed – Even professional responders cannot function properly
- Limited external support possible – Other communities are also affected, so they cannot help
- Examples: The 2004 Indian Ocean Tsunami, The 2010 Haiti Earthquake, The COVID-19 pandemic, A nuclear meltdown.
| Feature | EMERGENCY | DISASTER | CATASTROPHE |
|---|---|---|---|
| Community ability | CAN cope | CANNOT cope | Multiple communities CANNOT cope |
| Scope | Narrow (one building, one street) | Wide (one community or district) | Very wide (multiple communities, regions) |
| Onset | Predictable | Sudden, serious disruption | Massive, overwhelming |
| Response needed | Standard procedures | External support needed | External support limited or impossible |
| Examples | House fire, vehicle accident, single illness outbreak | Landslide in Bududa, flooding in Kasese, Ebola outbreak in one district | COVID-19 pandemic, massive earthquake affecting entire country |
| Nursing role | Standard care, may call for backup | Triage, coordination, request external help | Triage under extreme conditions, possibly working without supplies or support |
Memory Aid: "Emergency = We CAN handle it. Disaster = We CANNOT handle it alone. Catastrophe = NO ONE can handle it."
| Term | Simple Meaning | Nursing / Community Example |
|---|---|---|
| Vulnerability | Lack of capacity to deal with threats; can be physical, social, economic, or environmental | A village on a steep hillside with poorly constructed mud houses and no early warning system |
| Disaster Risk | Likelihood of hazards affecting people, property, and environment | Probability of landslide hitting Bududa district during rainy season |
| Acceptable Risk | Level of risk communities tolerate for daily activities | Flying in an airplane, eating street food, living in a mild earthquake zone |
| Residual Risk | Risk that remains AFTER all mitigation measures | House destroyed by earthquake despite building codes; flooding despite drainage |
| Hazard | Natural or human-made event threatening life or property | Dormant volcano, fault line, chemical factory near homes |
| Triage | Sorting victims by severity for treatment priority | Color tags (Red, Yellow, Green, Black) at mass casualty incident |
| Capacity | Community's ability to use resources to reduce risks | District having trained search and rescue teams, stocked medical supplies, evacuation routes |
| Capacity Building | Developing human skills to reduce risk | Training community health workers on first aid and early warning |
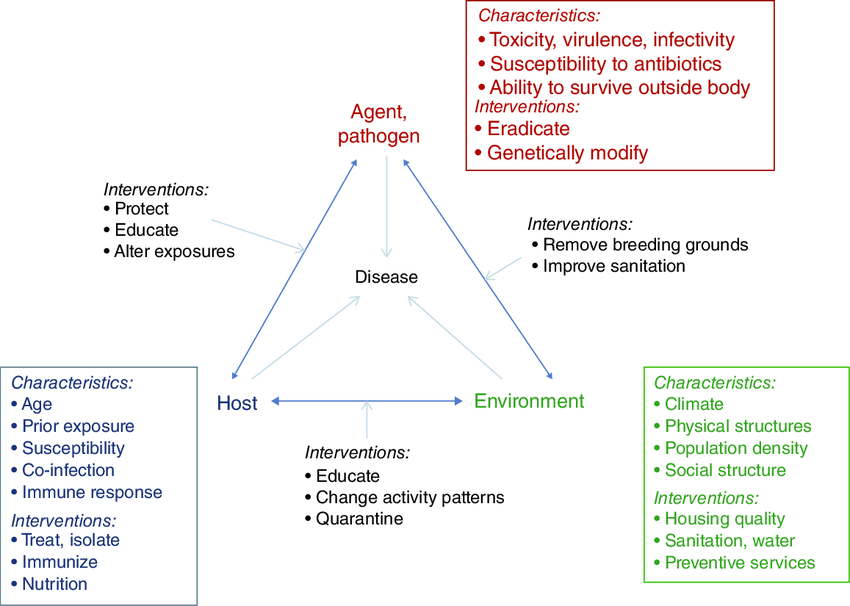
Definition: Epidemiology is the study of patterns of disease occurrence in human populations and the factors that influence these patterns.
In disaster nursing, epidemiology helps us understand WHO gets sick or injured, WHERE it happens, WHEN it happens, and WHY it happens.
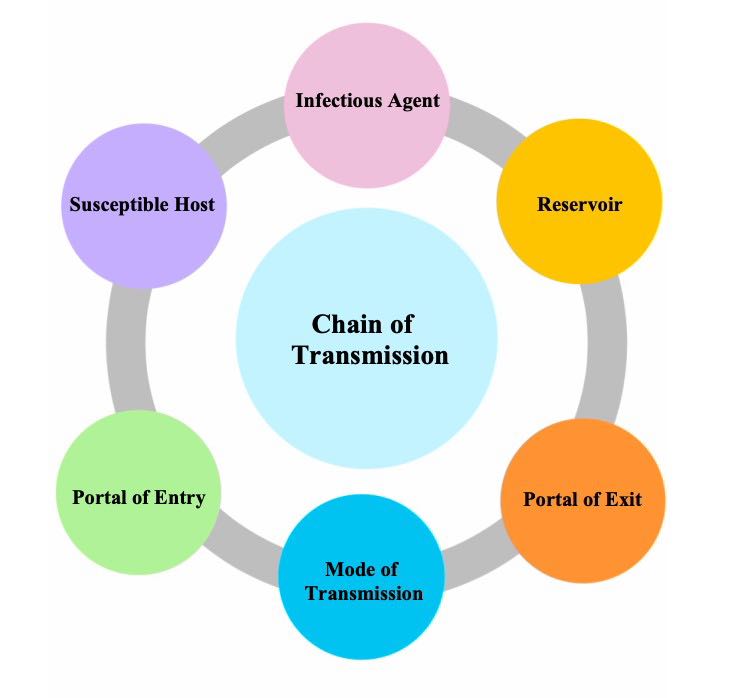
The Epidemiological Triad: In disaster epidemiology, we study three connected things:
- AGENT – The "what" that causes harm
- HOST – The "who" that is affected
- ENVIRONMENT – The "where" it happens
Definition: The agent is the physical, biological, or chemical entity that actually causes the injury or destruction.
Primary agents are the direct, immediate causes of injury or damage.
| Primary Agent | How It Causes Harm | Example |
|---|---|---|
| Falling objects | Hit people, cause trauma | Building collapse during earthquake |
| Building collapse | Crushing injuries, suffocation | Mud house collapsing in landslide |
| Heat | Burns, dehydration, heat stroke | Fire, volcanic eruption |
| Winds | Blow people away, throw objects, destroy structures | Cyclone, tornado |
| Water | Drowning, contamination, destruction of crops | Flood, tsunami |
Secondary agents are the indirect consequences that cause harm after the primary agent.
| Secondary Agent | How It Causes Harm | Example |
|---|---|---|
| Bacteria | Infection of wounds | Tetanus from dirty wounds after earthquake |
| Viruses | Disease outbreaks | Hepatitis E from contaminated water after flooding |
| Fungi | Skin infections, respiratory problems | Mold growing in flooded homes |
| Chemicals | Poisoning | Leaked fuel contaminating water supply |
Important Note: The primary agent causes the immediate disaster. The secondary agent causes the disaster AFTER the disaster. Nurses must be prepared for both.
Definition: The host refers to the characteristics of humans that influence how severely they are affected by a disaster.
| Factor | Explanation | Why It Matters |
|---|---|---|
| Age | Very young and very old people are more vulnerable | Children and elderly have weaker immune systems and less physical strength |
| Immune status | How well the body can fight infection | Malnourished people, HIV-positive people, and those with chronic diseases have weak immunity |
| Pre-existing health status | Current health conditions | A person with diabetes or hypertension will fare worse in a disaster |
| Degree of morbidity | How sick someone already is | Someone with tuberculosis is already struggling to survive |
| Emotional stability | Mental health and resilience | People with anxiety or depression may panic or become unable to make decisions |
| Pregnancy | Special physical needs | Pregnant women need more food, rest, and medical care; cannot move quickly |
| Nutritional status | Whether the person is well-fed | Malnourished children die faster from diarrhea or infections |
- Pregnant mothers: Need special care, cannot evacuate easily
- The elderly: Weak, may have chronic diseases, may live alone
- Children: Depend on adults, vulnerable to dehydration and malnutrition
- People with disabilities: May not be able to hear warnings, move, or communicate
- People with chronic diseases: Need regular medication (diabetes, HIV, hypertension)
- Malnourished individuals: Have no reserves to fight infection or survive trauma
Definition: Environmental factors are the conditions surrounding the host and agent that affect the outcome of a disaster.
| Physical Factor | How It Affects Disaster Outcome |
|---|---|
| Time of disaster | Disasters at night cause more deaths because people are sleeping and cannot see to escape |
| Weather conditions | Rain makes rescue harder; extreme heat causes dehydration |
| Water supply | Clean water prevents disease; contaminated water causes cholera and typhoid |
| Functionality of facilities | Working hospitals save lives; damaged hospitals cannot help |
| Communication systems | Working phones and radios allow warnings; broken systems leave people unaware |
| Roads and transportation | Good roads allow evacuation and supply delivery; destroyed roads trap people |
| Chemical Factor | Source | Effect on Humans |
|---|---|---|
| Contaminated groundwater | Leaking fuel tanks, industrial waste | Poisoning, cancer, birth defects |
| Contaminated food supply | Pesticides, spoiled food | Food poisoning, organ damage |
| Toxic fumes | Burning plastics, chemicals | Respiratory problems, death |
| Industrial chemicals | Factory leaks during earthquake | Burns, poisoning, long-term health effects |
| Biological Factor | Source | Disease Caused |
|---|---|---|
| Contaminated water | Sewage mixing with drinking water | Cholera, typhoid, dysentery |
| Improper waste disposal | Garbage attracting rats and flies | Plague, diarrhea, skin infections |
| Improper food storage | Food spoiling in heat without refrigeration | Food poisoning, salmonella |
| Vector breeding | Standing water after floods | Malaria (mosquitoes), dengue fever |
| Overcrowding | Many people in small shelters | Tuberculosis, meningitis, COVID-19 |
| Social Factor | How It Affects Recovery |
|---|---|
| Social support systems | People with family and friends recover faster; isolated people suffer more |
| Loss of family members | Grief and depression slow recovery; loss of breadwinner causes poverty |
| Changes in roles | When a father dies, a child may have to stop school to work; a mother may become head of household |
| Community cohesion | Strong communities help each other; divided communities fight over resources |
| Leadership | Good leaders organize relief; corrupt leaders steal aid |
| Cultural beliefs | Some beliefs may prevent people from seeking medical care or accepting help |
| The Agent (What) | Biological Agent: MRSA (Methicillin-resistant Staphylococcus aureus) bacteria |
| The Host (Who) | Elderly trauma patient with an open crush injury and compromised immune system from stress |
| The Environment (Where) | Overcrowded trauma ward, shortage of sterile gloves, overwhelmed nurses unable to perform hand hygiene |
| The Outcome | The chaotic environment allows MRSA to travel easily from nurse to patient. The bacteria enter the vulnerable host's open wound. The elderly patient's weak immune system cannot fight the infection. Result: Fatal sepsis. |
| The Agent (What) | Biological Agent: Vibrio cholerae (Cholera bacteria) |
| The Host (Who) | Severely malnourished child with low stomach acid and weak immunity |
| The Environment (Where) | Flooded clinic with no clean drinking water, overflowing latrines, dense population |
| The Outcome | Floodwater mixes sewage with drinking water. The contaminated water delivers a massive dose of cholera bacteria to the child. The malnourished host lacks immune reserves to fight it. Result: Rapid, severe dehydration and possible death. |
| The Agent (What) | Physical Agent: Toxic smoke inhalation and extreme heat |
| The Host (Who) | Bedbound ICU patient on mechanical ventilator (unable to flee) |
| The Environment (Where) | Hospital with failed fire alarms, blocked emergency exits, highly flammable oxygen tanks nearby |
| The Outcome | The unsafe structural environment traps the immobile host. The patient cannot move. The oxygen tanks fuel the fire. Result: Asphyxiation and death. |
Disasters do not happen by accident alone. They are caused by a combination of natural processes and human actions. Understanding causes helps nurses prevent disasters and prepare communities.
Explanation: The earth and atmosphere are always changing. When these changes become extreme, they cause disasters.
| Change | How It Causes Disaster | Ugandan Example |
|---|---|---|
| Extended drought | Lack of rain disturbs the water cycle; crops fail, animals die, people starve | Karamoja droughts causing famine and malnutrition |
| Excessive rainfall | Too much rain causes rivers to overflow and hillsides to collapse | Elgon region landslides during heavy rains |
| Temperature extremes | Extreme heat causes heat stroke and crop failure; extreme cold affects tropical crops | Unusually hot dry seasons affecting coffee yields |
| Tectonic movements | Earthquakes when plates shift | Occasional tremors in the Rwenzori region |
| Volcanic activity | Lava flows, ash clouds | Mt. Nyiragongo (near DRC border) affecting refugee camps |
Explanation: Poverty makes people vulnerable because they cannot afford safety. Poor people are forced to live in dangerous places and cannot build strong houses.
- Unsafe housing locations: Poor people settle on steep hills prone to landslides.
- Unsafe housing construction: Cannot afford cement and steel; build with mud and sticks.
- No savings for emergencies: When disaster strikes, they have no money to recover.
- No insurance: Lose everything with no compensation.
- Limited access to information: Cannot afford radios, phones, or televisions for warnings.
Ugandan Examples: Families in Kampala slums live in wetlands. Poor families in Bududa build on steep slopes. Karamoja pastoralists lose animals in drought because they have no alternative livelihood.
Explanation: When populations grow rapidly, more people compete for limited resources. This forces people into unsafe areas and can lead to conflict.
- Unsafe settlement: More people forced to live in floodplains, steep hillsides, and wetlands.
- Resource competition: Competition for land, water, and jobs leads to conflict.
- Environmental pressure: More farming, grazing, and logging degrade the environment.
- Overcrowding in cities: Slums grow with poor sanitation and unsafe housing.
- Crisis-induced migration: People flee conflict areas, creating refugee disasters.
Ugandan Examples: Kampala's population growth leading to massive slum expansion. Refugee influx from South Sudan and DRC straining resources. Competition for grazing land in Karamoja.
Explanation: Rapid urbanization happens when rural poor move to cities looking for jobs and security. Cities grow faster than infrastructure can support.
- Unplanned settlements: People build houses wherever they can, often in dangerous areas.
- Pressure on services: Water, sewage, and electricity systems cannot keep up.
- Environmental degradation: Wetlands filled, forests cut, hillsides built on.
- Increased disease risk: Overcrowding spreads tuberculosis, cholera, and COVID-19.
- Traffic and industrial accidents: More people and vehicles lead to more accidents.
Ugandan Examples: Kampala's wetlands were drained and built on. Boda-boda accidents are a leading cause of emergency department visits. Industrial areas with poor safety standards risk chemical spills.
Explanation: When communities change their traditional ways of doing things, sometimes the new ways are dangerous if not properly understood.
- New construction materials: Using cement and steel incorrectly because people are not trained.
- Abandoning traditional knowledge: Traditional building methods suited to local conditions are replaced by unsuitable modern methods.
- New farming practices: Introducing crops that deplete soil or need too much water.
- Changing social structures: Traditional community support systems break down.
Examples: Building with concrete blocks but not using proper foundations. Abandoning terracing on hillsides. Replacing drought-resistant traditional crops.
Explanation: Environmental degradation means damaging the natural environment that protects us from disasters.
| Type | Cause | Disaster Result |
|---|---|---|
| Deforestation | Cutting trees for charcoal and farming | Landslides, soil erosion, loss of water sources |
| Overgrazing | Too many animals eating grass | Desertification, dust storms, famine |
| Poor cropping patterns | Planting same crop repeatedly without rest | Soil depletion, lower yields, food insecurity |
| Topsoil stripping | Removing the fertile top layer of soil | Cannot grow crops, mudslides |
| Water depletion | Using groundwater faster than it replenishes | Wells dry up, drought worsens |
| Wetland destruction | Draining wetlands for building | Worse flooding, loss of water purification |
| Pollution | Dumping chemicals and waste | Contaminated water, disease outbreaks |
Ugandan Examples: Deforestation on Mt. Elgon slopes. Overgrazing in Karamoja. Wetland destruction in Kampala causing floods.
Explanation: When people do not know about risks, protective measures, safe locations, or evacuation procedures, they cannot protect themselves.
- Do not know the risk: People build in floodplains because they do not know floods happen there.
- Do not know warning signs: Communities miss landslide warnings (cracks in ground, tilting trees).
- Do not know evacuation routes: People panic and run the wrong way.
- Do not know first aid: Simple injuries become fatal because no one knows basic care.
- Do not know where to get help: People suffer needlessly because they do not know about available services.
Ugandan Examples: Communities in landslide-prone areas not recognizing ground cracks. People not knowing stagnant water breeds malaria mosquitoes.
Explanation: Human conflict is a major cause of disasters. War destroys infrastructure, displaces populations, and creates health crises.
Causes: Competition for scarce resources, religious/ethnic intolerance, ideological differences, colonial legacy.
| Effect | Health Impact |
|---|---|
| Destruction of hospitals and clinics | No access to healthcare |
| Displacement of populations | Refugee camps with overcrowding and disease |
| Food supply disruption | Malnutrition and famine |
| Breakdown of water and sanitation | Cholera, typhoid, dysentery |
| Mental health trauma | PTSD, depression, anxiety |
| Sexual violence | Physical injury, HIV transmission, psychological trauma |
Ugandan Examples: Rwandan Genocide (1994) massive refugee influx into Uganda. LRA conflict in Northern Uganda. South Sudan conflict refugee crisis in West Nile. Karamoja cattle raids.
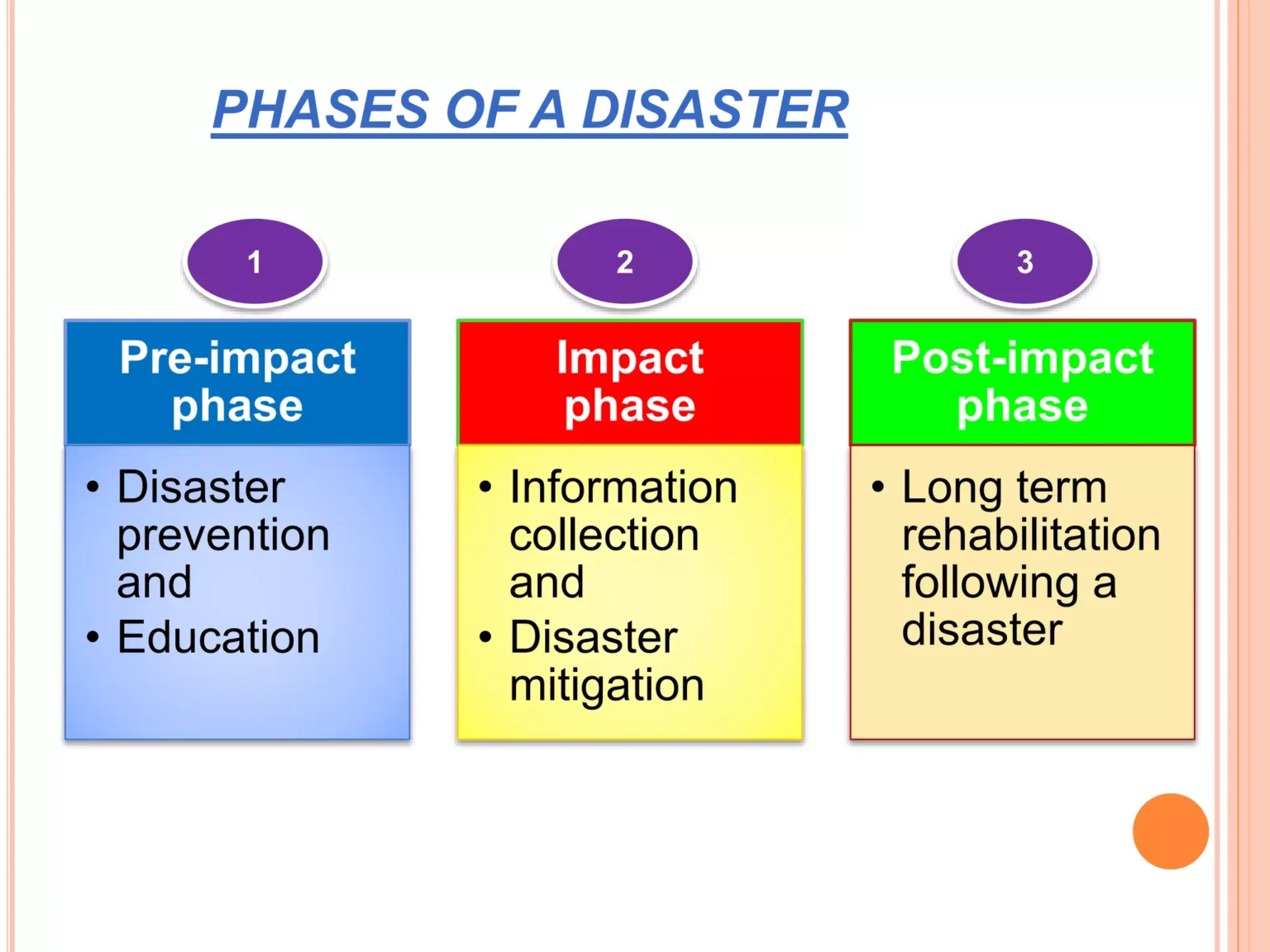
Every disaster goes through three phases. The actions that nurses and emergency personnel take depend on which phase the disaster is in.
Definition: The pre-impact phase is the initial phase before the disaster actually occurs. It is the time when warning signs appear and preparation happens.
- Warning is given at the first sign of possible danger
- This is the time for preparedness planning
- Emergency supplies are organized, and communication systems are tested
- Communities are educated
Why the Earliest Possible Warning is Crucial: Prevents loss of life, minimizes damage, allows preparation, and reduces panic.
| Activity | Details | Nursing Role |
|---|---|---|
| Emergency preparedness planning | Creating disaster response plans | Participate in planning committees; know the hospital disaster plan |
| Opening first aid centers | Setting up emergency treatment areas | Help set up and stock first aid stations |
| Communication | Ensuring radios, phones, and warning systems work | Test communication equipment; establish contact networks |
| Community education | Teaching people what to do | Conduct community sensitization; teach first aid and evacuation |
| Preparing emergency shelters | Identifying and setting up safe buildings | Help prepare shelters; ensure medical supplies are available |
| Stocking medical equipment | Gathering medicines, bandages, equipment | Inventory supplies; request additional stock |
| Training drills | Practicing disaster response | Participate in and help organize drills |
- Sensitize the community: Go door-to-door, teach warning signs.
- Assist in making emergency shelters: Identify safe buildings, set up sleeping/sanitation areas, ensure accessibility.
- Prepare medical equipment: Check emergency drug stocks, prepare first aid kits, test generators.
- Educate the community: Teach family disaster plans, food/water storage, basic first aid, and evacuation routes.
"Pre-impact = PREPARE. It is the time to PLAN, EDUCATE, and STOCK SUPPLIES."
Definition: The impact phase occurs when the disaster has actually happened. It is the time of enduring hardship, injury, and trying to survive.
- The disaster is happening or has just happened
- It is an emergency period where people help neighbors
- It is a time of "holding on" until outside help arrives
| Activity | Details | Nursing Role |
|---|---|---|
| Preliminary assessment | Determining nature, extent, and area of disaster | Rapidly assess number and types of injuries |
| Needs assessment | Identifying what victims need | Assess health needs of the community |
| Disaster health services assessment | Determining what medical services are needed | Identify type and number of health services required |
| Reporting | Informing authorities | Report to disease control centers and take action |
| Triage | Sorting victims by severity | Use color-coded triage system |
| Treatment | Providing immediate medical care | Treat injured persons; stabilize critical patients |
| Search and rescue | Finding and helping trapped people | Coordinate with rescue teams; provide medical support |
| Reunion activities | Helping families find each other | Maintain records; help reunite separated families |
- Assess health needs: Walk through affected areas, count injured/dead, identify immediate health threats.
- Provide physical support: Administer first aid, set up IV fluids, control bleeding, immobilize fractures, manage pain.
- Provide psycho-social support: Comfort mothers/children, reassure frightened patients, listen to fears.
- Special care for vulnerable groups: Set up special shelters, ensure children are protected, prioritize elderly/disabled.
- Coordinate search and rescue: Work with police/volunteers, provide medical support, triage victims immediately.
- Reunion activities: Keep records of admitted patients, help trace missing family members.
Victims of disaster usually go through four stages of emotional response. Nurses must recognize these stages to provide appropriate support.
- Denial Stage
- Description: Victims deny the magnitude of the problem or seem unaffected emotionally.
- Why It Happens: The mind protects itself from overwhelming shock (a temporary defense mechanism).
- Nursing Care: Do not force reality, stay with the person, provide simple information, ensure physical safety.
- Strong Emotional Response
- Description: The person becomes aware of the disaster but regards it as overwhelming. Emotions flood out.
- Common Reactions: Fighting/fleeing, weeping, stammering, trembling, sadness, anger, confusion.
- Nursing Care: Stay calm, listen without judgment, provide physical comfort, use simple/soothing words, do not leave them alone.
- Acceptance Stage
- Description: Victims begin to accept what has happened and are ready to move forward.
- Characteristics: Able to think more clearly, willing to make decisions, open to help.
- Nursing Care: Help develop decision-making skills, encourage hope, involve them in their care, connect them with resources.
- Recovery Stage
- Description: Victims recover from crisis reaction, feeling they are back to normal.
- Characteristics: Returns to daily activities, re-establishes relationships, feels hopeful.
- Nursing Care: Resettle victims, discuss empowerment, support income generation, continue mental health support, celebrate progress.
Definition: The post-impact phase is the period of recovery from the emergency phase. It ends when normal community order and functioning are restored.
- The immediate danger has passed, recovery and rebuilding begin.
- This phase may last months or even years.
- Long-term care and support are needed; communities must be empowered.
| Activity | Details | Nursing Role |
|---|---|---|
| Counseling | Long-term mental health support | Provide ongoing psychological support; refer severe cases |
| Rehabilitation | Physical and social recovery | Start rehabilitation programs; physiotherapy, prosthetics |
| Community sensitization | Educating for future preparedness | Teach lessons learned; improve early warning systems |
| Empowerment | Helping communities help themselves | Support income-generating activities |
| Reconstruction | Rebuilding infrastructure | Advocate for safe building practices |
| Monitoring health | Watching for delayed health effects | Monitor for disease outbreaks, malnutrition, mental health issues |
Disaster management follows 8 core principles. These guide all actions during a disaster.
Full Meaning: O = Occurrence prevention.
Explanation: The primary goal is to prevent disasters whenever possible. This involves proactive measures and risk reduction strategies to avoid the disaster happening in the first place.
- Building dams and levees (Prevents flooding)
- Enforcing building codes (Prevents collapse)
- Reforestation (Prevents landslides)
- Vaccination campaigns (Prevents outbreaks)
Nursing Role: Advocate for prevention policies, teach communities, participate in vaccination programs.
Full Meaning: N = Number of casualties limited.
Explanation: If prevention is not possible, the focus shifts to minimizing casualties. Quick and effective responses are implemented to reduce the impact on human lives.
Nursing Role: Triage efficiently, provide rapid first aid, help organize evacuations, communicate clearly.
Full Meaning: E = Evaluate personnel.
Explanation: Medical personnel are the health providers. If they are injured, they cannot help others. Their well-being must be checked first so they can continue saving lives.
Nursing Role: Check on colleagues after a disaster, report injuries immediately, accept help when you are injured, practice self-care.
Full Meaning: F = First aid provision.
Explanation: Rapid and efficient first aid is crucial to address immediate medical needs, stabilize victims, and prevent further harm.
Nursing Role: Always carry a first aid kit, be prepared to provide care anywhere, train community members, prioritize life-threatening conditions (bleeding, airway, shock).
Full Meaning: F = Further casualties prevented.
Explanation: After the initial impact, ongoing efforts are made to prevent additional casualties. This involves continuous risk assessment and safety measures.
Nursing Role: Continuously assess for new dangers, implement infection control measures, educate displaced people about safety, report new hazards.
Full Meaning: R = Rescue and search.
Explanation: Swift rescue operations are essential to saving lives. This includes searching for and rescuing victims as quickly as possible while minimizing additional damage.
Nursing Role: Provide medical support at rescue sites, triage rescued victims immediately, stabilize patients for transport, support rescue workers.
Full Meaning: R = Reconstruction.
Explanation: For those severely affected, the focus shifts to reconstruction and rehabilitation. This principle emphasizes restoring a meaningful life for disabled casualties.
Nursing Role: Coordinate rehabilitation services, advocate for disabled-friendly facilities, support vocational training, provide long-term psychological support.
Full Meaning: M = Medical services continuous.
Explanation: Medical services are sustained at the disaster scene, and severely injured victims are promptly transported to hospitals for advanced care.
Nursing Role: Set up treatment areas, ensure staffing rotations, monitor patients continuously, arrange/prioritize transport to hospitals.
| Letter | Principle | Simple Meaning |
|---|---|---|
| O | Occurrence prevention | Stop it before it starts |
| N | Number of casualties limited | Save as many lives as possible |
| E | Evaluate personnel | Check if medics are okay first |
| F | First aid provision | Immediate medical care |
| F | Further casualties prevented | Stop the domino effect |
| R | Rescue and search | Find and save victims |
| R | Reconstruction | Rehabilitate the disabled |
| M | Medical services continuous | Never stop treating patients |
Q1: Differentiate between an emergency and a disaster.
Answer: An emergency is a situation the community IS CAPABLE of coping with using its own resources. A disaster is a situation the community is INCAPABLE of coping with and requires external support.
Q2: List the four types of vulnerability and give one example of each.
Answer: Physical: Wooden homes are vulnerable to fire. Social: Children and elderly cannot evacuate during floods. Economic: Poor families live in squatter settlements in unsafe areas. Environmental: Wetlands are sensitive to pollution and salinity.
Q3: What is the epidemiological triad in disaster management?
Answer: The three components are: Agent (The physical entity causing harm), Host (The human characteristics affecting outcome), Environment (The surrounding conditions).
Q4: Describe the three phases of disaster action.
Answer: Pre-impact (Before disaster; warning and preparation), Impact (During disaster; survival and immediate response), Post-impact (After disaster; recovery and rehabilitation).
Q5: List the four stages of emotional response to disaster.
Answer: Denial stage, Strong emotional response, Acceptance stage, Recovery stage.
Q6: Using the mnemonic ONEFFRRM, list the 8 principles of disaster management.
Answer: Occurrence prevention, Number of casualties limited, Evaluate personnel, First aid provision, Further casualties prevented, Rescue and search, Reconstruction, Medical services continuous.
You are a community health nurse in Bududa. Heavy rains have been falling for three days. You notice cracks appearing in the ground behind several houses.
- Which disaster phase is this? Pre-impact.
- What is your immediate role? Warn community, help evacuate, contact authorities.
- Which type of vulnerability is most relevant? Physical and environmental.
- What should you teach the community? Warning signs, evacuation routes, emergency kit preparation.
After heavy rains, the Nyamwamba River has burst its banks. Hundreds of people are displaced. You are working at a temporary health center in a school.
- Which disaster phase is this? Impact.
- What are your nursing priorities? Triage, treat injuries, prevent cholera, provide psycho-social support.
- Which secondary agents should you watch for? Cholera, typhoid, malaria.
- How do you prevent further casualties? Ensure clean water, proper sanitation, mosquito control.
You are a nurse at a regional referral hospital. A patient arrives with symptoms of Ebola. The hospital is not prepared.
- Is this an emergency or disaster? Potentially a disaster if the hospital cannot cope.
- What is your first action? Isolate the patient, notify authorities, protect yourself with PPE.
- Which principle of disaster management applies first? Occurrence prevention – prevent spread.
- How do you protect medical personnel? Evaluate personnel – ensure they are not exposed, provide PPE.
- Disaster = Community CANNOT cope alone.
- Vulnerability has four types: Physical, Social, Economic, Environmental.
- Risk = Likelihood + Consequences.
- Residual risk always remains even after prevention.
- Triage saves the maximum number of lives with limited resources.
- The Epidemiological Triad = Agent + Host + Environment.
- Disasters have three phases: Pre-impact, Impact, Post-impact.
- Emotional stages: Denial → Strong emotion → Acceptance → Recovery.
- ONEFFRRM = The 8 principles of disaster management.
- Nurses play roles in ALL phases: preparation, response, and recovery.
| Phase | Key Nursing Roles |
|---|---|
| Pre-impact | Community education, shelter preparation, stock supplies, sensitize |
| Impact | Triage, first aid, psycho-social support, special care for vulnerable, search and rescue support |
| Post-impact | Counseling, rehabilitation, empowerment, income generation support, long-term monitoring |
| Group | Special Needs | Nursing Action |
|---|---|---|
| Pregnant women | Antenatal care, safe delivery, nutrition | Prioritize for shelter, ensure clean delivery kits, monitor for complications |
| Children | Nutrition, hydration, protection | ORS, immunization, child-friendly spaces, tracing separated children |
| Elderly | Mobility assistance, chronic disease care | Help with evacuation, ensure medications available, prevent falls |
| Disabled persons | Accessible evacuation, communication aids | Plan accessible routes, use visual/tactile warnings, assign helpers |
| Chronic disease patients | Regular medication, monitoring | Stock essential medicines (insulin, hypertension drugs, ARVs), ensure continuity of care |
- 📌 Know your definitions word-for-word – Examiners love exact definitions from WHO.
- 📌 Be able to give Ugandan examples – This shows you understand the local context.
- 📌 Practice the ONEFFRRM mnemonic – It is guaranteed to appear on exams.
- 📌 Understand the difference between emergency, disaster, and catastrophe – This is a classic exam question.
- 📌 Know the four emotional stages – Mental health in disasters is increasingly tested.
- 📌 Be ready to discuss nursing roles – You must know what YOU as a nurse would do in each phase.
- World Health Organization (WHO). (2020). Health Emergency and Disaster Risk Management Framework. Geneva: WHO.
- Veenema, T. G. (2018). Disaster Nursing and Emergency Preparedness (4th ed.). Springer Publishing Company.
- International Council of Nurses (ICN). (2019). Core Competencies in Disaster Nursing Version 2.0.
- Ministry of Health, Uganda. National Guidelines for Emergency Medical Services and Disaster Preparedness.
- Uganda Office of the Prime Minister (OPM). National Policy for Disaster Preparedness and Management.
Introduction to Disaster in Nursing Read More »