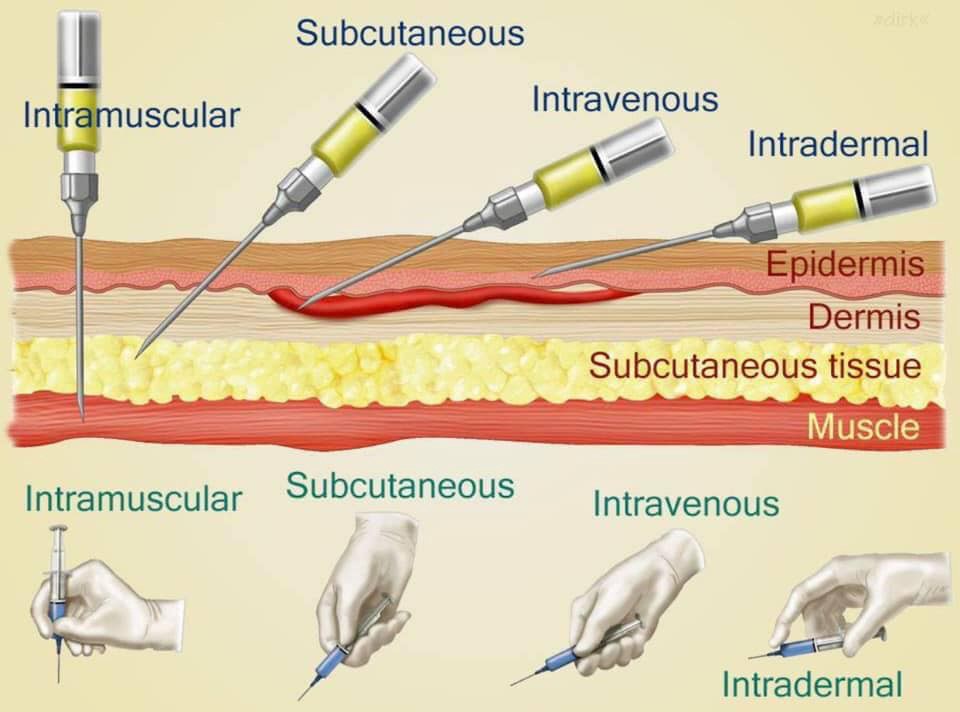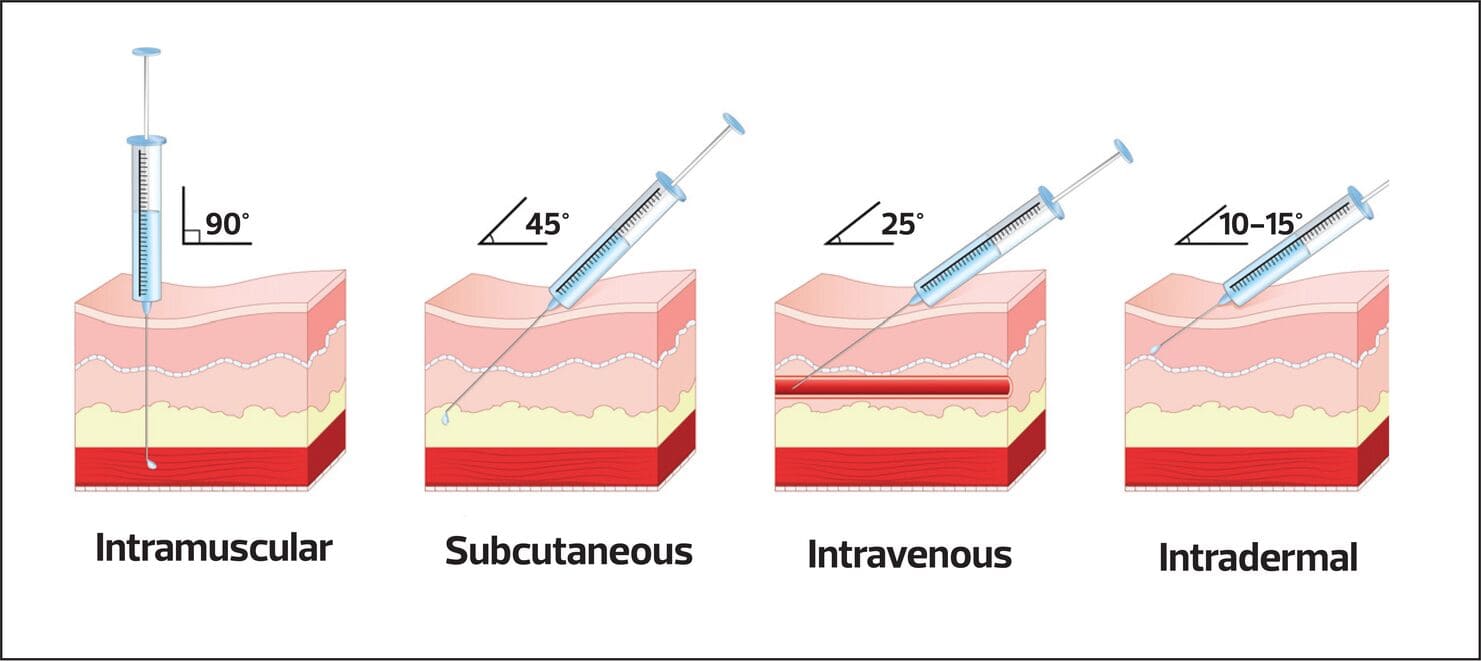
Hyperthyroidism
The thyroid gland is a crucial and highly vascular component of the endocrine system. It is located at the anterior (front) of the neck, situated just below the larynx (Adam's apple) and wrapping around the anterior and lateral surfaces of the trachea (windpipe).
- Shape & Structure: It is classically described as butterfly-shaped, consisting of a right and left lobe connected by a narrow band of tissue called the isthmus.
- Physical Characteristics: The gland weighs between 20 and 60 grams on average in adults. It has a distinctive brownish-red color due to its incredibly rich blood supply (highly vascularized).
The thyroid gland produces three principle hormones that exert systemic effects:
- Thyroxine (T4): The primary prohormone secreted by the gland.
- Triiodothyronine (T3): The highly active, potent form of the hormone.
- Calcitonin: Produced by the parafollicular cells (C-cells), responsible for calcium homeostasis (it lowers blood calcium levels by inhibiting osteoclast activity).
Iodine is an absolute elemental requirement for the synthesis of thyroid hormones. Without adequate dietary iodine, the gland cannot synthesize T3 or T4, often leading to compensatory hypertrophy (goiter).
- Uptake of Iodine: Active transport of iodide from the blood into the follicular cells.
- Formation of Active Iodine: Oxidation of iodide to active iodine.
- Thyroglobulin Synthesis: Binding of iodine to tyrosine residues on the thyroglobulin molecule, leading to the coupling and synthesis of T3 and T4.
Regulation operates strictly on a negative feedback loop. The Hypothalamus releases Thyrotropin-Releasing Hormone (TRH) → stimulates the Anterior Pituitary to release Thyroid-Stimulating Hormone (TSH) → stimulates the Thyroid Gland to synthesize and release T3 and T4. High circulating levels of T3/T4 will subsequently inhibit the release of both TRH and TSH.
Thyroid hormones affect virtually every cell and all the organs of the human body. Their primary role is the modulation of the Basal Metabolic Rate (BMR).
- Metabolism & Weight: Regulates the exact rate at which calories are burned, profoundly affecting weight loss or weight gain.
- Cardiovascular: Can speed up or slow down the heartbeat, altering cardiac output.
- Thermoregulation: Can raise or lower core body temperature.
- Gastrointestinal: Influences the rate at which food moves through the digestive tract (peristalsis).
- Musculoskeletal: Controls the way muscles contract and the rate at which dying cells are replaced (bone and tissue turnover).
- Growth & Development: Crucial in children for physical growth and proper maturation of the brain.
- Neurological: Activation of the nervous system leads to improved concentration and faster reflexes.
Hyperthyroidism is the clinical condition that occurs due to the excessive production of thyroid hormone by the thyroid gland. Alternatively, it is defined as the hyperactivity of the thyroid gland leading to a sustained increase and excessive synthesis and release of thyroid hormones, resulting in an accelerated hypermetabolic state in the peripheral tissues.
Incidence: It is the second most common thyroid problem (behind hypothyroidism). It exhibits a strong gender predilection, affecting women eight times more frequently than men. The peak onset typically occurs between the ages of 20 and 40 years.
| Type | Description & Pathophysiology |
|---|---|
| 1. Primary Hyperthyroidism | Arising directly from an intrinsic thyroid abnormality (the gland itself is at fault, autonomously overproducing hormones). |
| 2. Secondary Hyperthyroidism | Arising from a process outside of the thyroid gland, such as a TSH-secreting pituitary tumor driving an otherwise healthy thyroid to overproduce. |
| 3. Apathetic Hyperthyroidism |
A highly deceptive, atypical presentation occurring primarily in the elderly (>70 years, though possible at any age). Key Features: The typical hallmark features of thyroid hormone excess seen in younger patients (tachycardia, hyperactivity, prominent tremors) are completely blunted. The cardinal symptoms are instead depression and apathy. Clinical Note: The absence of classical signs severely delays diagnosis. It is often misdiagnosed as a primary psychiatric disorder, dementia, or occult malignancy. Diagnosis is typically made incidentally during investigations for unexplained weight loss or worsening cardiovascular disease (e.g., new-onset atrial fibrillation). |
- A family history of thyroid disease, particularly of Graves' disease.
- Female sex.
- A personal history of certain chronic autoimmune illnesses, such as Type 1 Diabetes Mellitus, Pernicious Anemia (Vitamin B12 deficiency), or Primary Adrenal Insufficiency (Addison's disease).
- Advanced age (Over the age of 60).
- Consuming an iodine-rich diet or taking medications containing high concentrations of iodine (e.g., Amiodarone, an antiarrhythmic drug).
- Prior history of thyroid surgery or a pre-existing thyroid problem such as a goiter (swollen thyroid gland).
- Exposure to radiation therapy, which may predispose a patient to develop hyperthyroidism or thyroid nodules.
- Smoking: Women who smoke have nearly double the risk of developing Graves' disease compared to non-smokers.
- Graves' Disease: The absolute most common cause, accounting for 50-80% of cases worldwide. It is an autoimmune disorder characterized by the presence of Thyroid-Stimulating Immunoglobulins (TSIs) that bind to TSH receptors, constantly stimulating the gland. Classic triad: Hyperthyroidism, Diffuse Goiter, and Exophthalmos (bulging eyes).
- Toxic Thyroid Adenoma: A benign, solitary tumor (nodule) in the thyroid that produces thyroid hormones independently of TSH control.
- Toxic Multinodular Goiter (Plummer's Disease): If there is more than one functioning, autonomous nodule, it is termed a multinodular goiter. It occurs more commonly in the elderly, especially those with a long-standing history of goiter.
- Thyroiditis: Inflammation of the thyroid gland (viral, autoimmune, or post-partum) which causes pre-formed T4 and T3 to "leak" out of the damaged gland and into the bloodstream. Often causes a transient hyperthyroidism followed by hypothyroidism.
- Excessive intake of thyroid hormone: Factitious hyperthyroidism or iatrogenic overdose of levothyroxine medication.
- Pituitary Tumors: An adenoma in the pituitary gland may produce an abnormally high, unregulated secretion of TSH, driving secondary hyperthyroidism.
- Excessive Iodine Intake: Can trigger hyperthyroidism in patients with pre-existing endemic goiters (Jod-Basedow phenomenon).
- Struma Ovarii: A very rare form of monodermal ovarian teratoma that contains mostly functioning thyroid tissue, leading to ectopic hyperthyroidism.
The pathophysiology centers around a dramatic acceleration of metabolic processes. Graves' disease may be due to excessive stimulation of the adrenergic nervous system or direct effects of excessive levels of circulating Thyroid Hormone (TH).
- Hyperthyroidism is characterized by a complete loss of the normal regulatory controls (negative feedback fails).
- Because the action of TH on the body is stimulatory, hypermetabolism results alongside significantly increased sympathetic nervous system activity.
- Excessive amounts of TH forcefully stimulate the cardiac system, increase the number and sensitivity of beta-adrenergic receptors (responsiveness), and increase peripheral blood flow.
- Metabolism increases so greatly that it leads to a negative nitrogen balance, lipid depletion, a state of profound nutritional deficiency, and rapid weight loss despite increased appetite.
- It also results in the altered secretion and metabolism of hypothalamic, pituitary, and gonadal hormones. If hyperthyroidism occurs before puberty, sexual development is notably delayed in both genders.
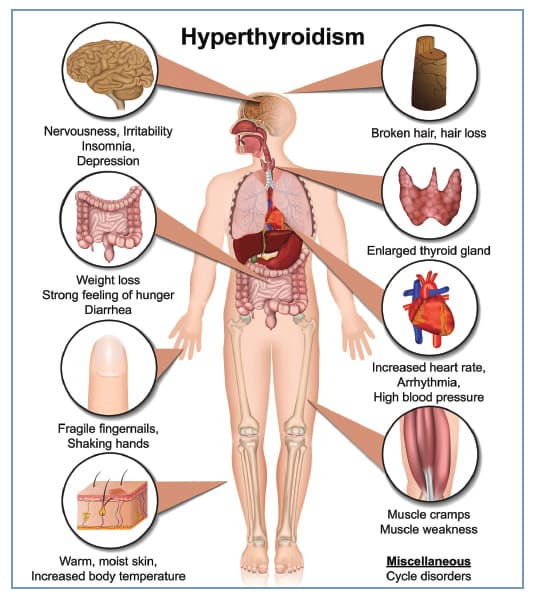
The systemic hypermetabolic state produces a vast array of symptoms across nearly every bodily system.
| Body System | Signs & Symptoms |
|---|---|
| 1. Cardiovascular |
|
| 2. Respiratory |
|
| 3. Gastrointestinal |
|
| 4. Integumentary |
|
| 5. Musculoskeletal |
|
| 6. Nervous System |
|
| 7. Reproductive |
|
| 8. Ophthalmic (Graves' Specific) |
|
- History Taking: Inquire about family history, geographical area (iodine availability), dietary patterns, current medications (especially amiodarone or supplements), and classic signs (heat intolerance, increased sweating, weight loss).
- Physical Examination: Assess vital signs (tachycardia, elevated BP), weigh the patient, inspect for hand tremors, examine the eyes for exophthalmos, and palpate the neck for an enlarged thyroid gland (goiter) or nodules. Note the presence of warm, moist skin.
- Laboratory Tests:
- Classic Profile: Increase in serum T4 and T3, and a profound decrease in TSH (in primary hyperthyroidism).
- Measurements of Free Tri-iodothyronine (Free T3) and Free Thyroxine (Free T4).
- Measurements of Total T3 and Total T4.
- Cholesterol Level (usually abnormally low due to hypermetabolism).
- Antibody Test: TSH-receptor antibodies (TRAb) or Thyroid-stimulating immunoglobulins (TSI) confirm Graves' disease.
- Basal Metabolic Rate (BMR) - elevated.
- Radiologic Exams: Ultrasound (assesses gland size and nodules), Thyroid Scan (evaluates structure), Iodine uptake scan (differentiates Graves' vs. Thyroiditis based on high vs. low radioactive iodine uptake).
- Histologic Exam: Fine Needle Aspiration (FNA) Thyroid biopsy to rule out malignancy in nodules.
- Ophthalmological Examination: To formally determine the extent of exophthalmos and optic nerve involvement.
Management strictly depends on the exact cause, the age of the patient, the severity of the disease, and existing complications. The ultimate goal of therapy is to bring the metabolic rate to normal (a euthyroid state) as safely and as soon as possible. Three primary forms of treatment are available:
- Antithyroid Medications (Thionamides): Drugs such as Carbimazole, Methimazole (Tapazole), and Propylthiouracil (PTU).
Mechanism: They directly inhibit the synthesis of new thyroid hormones in the gland.
Limitation: They do not destroy pre-formed circulating hormone, so they may take several weeks to months to become fully effective. Treatment must often be continued for 12-18 months. - Beta-Blockers: Drugs such as Propranolol or Metoprolol.
Mechanism: These medications do not treat the underlying levels of thyroid hormone. Instead, they block the sympathetic nervous system overdrive, providing rapid symptomatic relief from anxiety, severe shaking (tremors), and dangerous tachycardias.
This is often the treatment of choice for non-pregnant adults.
- A specifically calculated small amount of radioactive iodine is taken orally.
- The overactive thyroid cells preferentially absorb it, and the localized radiation destroys the hyperactive tissue.
- This causes the thyroid gland to physically shrink and forces the levels of thyroid hormone to go down.
- Clinical Consideration: It almost always eventually causes hypothyroidism. However, from a medical standpoint, hypothyroidism is vastly easier and safer to manage (requiring just a once-a-day levothyroxine supplement) than life-threatening hyperthyroidism.
Surgical intervention involves Subtotal Thyroidectomy (leaving a small portion behind) or Total Thyroidectomy (entire gland removed). It is not extensively used as the first-line treatment because radioactive iodine is highly effective and non-invasive. Furthermore, surgery carries risks of bleeding, cutting the recurrent laryngeal nerve (making swallowing/speaking difficult), and inadvertently removing the parathyroid glands.
Indications for Surgery include:
- A massive goiter causing tracheal compression or choking.
- Patients who have been wholly unresponsive to or are allergic to antithyroid therapy.
- Suspicion or confirmation of thyroid cancer.
- Patients unresponsive to radiotherapy or pregnant women who cannot undergo radiation.
Endoscopic Thyroidectomy: A minimally invasive procedure where several small incisions are made and a scope is inserted to remove tissue. It is appropriate for patients with small nodules (less than 3 cm) and absolutely no evidence of malignancy. Advantages over open surgery include significantly less scarring, reduced pain, and a faster return to normal activity.
This is a supreme medical emergency. It is an acute, severe, and rare condition that occurs when massive, excessive amounts of thyroid hormones are dumped into the circulation. It is often triggered by stressors such as infection, trauma, or surgery in a patient with poorly controlled hyperthyroidism. If left untreated, it is usually fatal. With aggressive, proper treatment, the mortality rate is reduced substantially.
- Symptoms Include:
- Dangerously high fever (above 38.5°C / 101.3°F, sometimes reaching 105°F+).
- Extreme tachycardia (heart rate can exceed 130-200 bpm).
- Severely altered neurologic or mental state (agitation, delirium, psychosis, coma).
- Massively exaggerated symptoms of baseline hyperthyroidism (severe vomiting, diarrhea, dehydration).
- Management of Thyroid Storm:
- Treatment is first and foremost directed toward relieving the immediate life-threatening symptoms.
- Acetaminophen is given aggressively for the fever.
- CRITICAL RULE: Aspirin is completely avoided. Aspirin chemically binds with the same serum transport proteins as T4, violently displacing the bound hormone and freeing additional, highly active T4 into the circulation, actively worsening the crisis.
- Intravenous fluids (to combat dehydration from diarrhea/diaphoresis) and a cooling blanket may be ordered to forcefully cool the patient.
- A Beta-adrenergic blocker, such as intravenous propranolol, is given immediately for tachycardia, arrhythmia, and sympathetic symptom control.
- Oxygen is administered, and the head of the bed is elevated because the extreme metabolic rate massively increases the body's cellular oxygen requirements.
- Once the life-threatening symptoms are stabilized and the patient is safe, the underlying thyroid dysfunction is definitively treated (PTU, iodine, steroids).
- General: Heart problems (fast heart rate, abnormal rhythm like A-fib, heart failure), Osteoporosis, progressive Airway Obstruction (from enlarging goiter), and severe Respiratory distress. Also, eventual Hypothyroidism (underactive thyroid) following treatment.
- Surgery-Related Complications:
- Immediate: Hemorrhage (life-threatening if it compresses the airway).
- Short Term: Infection.
- Long Term: Scarring of the neck, Hoarseness due to permanent damage to the recurrent laryngeal nerve (voice box), and Hypocalcemia/Hypoparathyroidism due to the inadvertent damage or removal of the parathyroid glands.
- Monitor the patient closely and continuously until normal thyroid activity is fully restored.
- Monitor vital signs relentlessly. Immediately report any increases in pulse or blood pressure to the registered nurse or physician, as this signals impending crisis.
- Monitor lung sounds carefully because crackles can indicate the onset of high-output heart failure.
- Assess the level of anxiety and the patient's ability to cope with symptoms. Monitor weight trends, bowel function, and sleep patterns.
- Assess the eyes for risk for injury caused by exophthalmos, and carefully note the degree of any muscle weakness.
- CRUCIAL WARNING: Never palpate the thyroid gland of a patient with active hyperthyroidism. Palpation or rough handling can physically squeeze the gland, stimulating a massive release of thyroid hormone and precipitating a deadly thyrotoxic crisis.
| No. | Diagnosis & Interventions | Rationale / Expected Outcome |
|---|---|---|
| 1. Hyperthermia related to hypermetabolic state as evidenced by elevated temperature. | ||
| 1 | Monitor temperature closely. Administer acetaminophen as ordered. AVOID ASPIRIN. | Aspirin increases free active circulating thyroid hormone. Acetaminophen safely reduces temperature. |
| 2 | Apply cooling blanket as ordered. Set it to 1-2 degrees below current temp. Wrap extremities with towels. | External cooling combats the hypermetabolism. Wrapping extremities prevents shivering, which would counterproductively increase body heat. |
| 3 | Offer copious oral or IV fluids. | Replaces massive fluid volumes lost through profound diaphoresis. Expected Outcome: Patient's body temperature will remain within normal limits. |
| 2. Diarrhea related to an increase in peristalsis as evidenced by frequent loose stools. | ||
| 4 | Provide a strict low-fiber diet and small, frequent meals of bland foods (e.g., bananas, rice, applesauce, toast - BRAT diet). | High fiber heavily stimulates peristalsis. Bland foods are easily digested and less likely to worsen diarrhea. |
| 5 | Monitor electrolytes (especially sodium and potassium) and watch for severe dehydration. | Chronic diarrhea forces the rapid loss of critical electrolytes and fluid volume. |
| 6 | Keep the perianal skin clean and dry; generously apply barrier cream. | Protects delicate skin from caustic breakdown due to frequent stooling. Expected Outcome: Maintain fluid and electrolyte balance. |
| 3. Inadequate protein energy intake related to profoundly increased metabolism. | ||
| 7 | Consult dietician for a high-calorie diet (4000 to 5000 calories/day) with high protein, split into six meals per day. | Massive caloric intake is mandatory to compensate for the hypermetabolic state and prevent severe negative nitrogen balance and muscle wasting. |
| 8 | Determine healthy weight for height, monitor weight strictly weekly. Monitor IV infusions, skin turgor, and vital signs. | Tracks the effectiveness of nutritional interventions and overall hydration status. Expected Outcome: Patient will have stable weight proportional to height. |
| 4. Disrupted Sleep Pattern related to sympathetic stimulation as evidenced by difficulty sleeping. | ||
| 9 | Provide a quiet, restful, calm environment. Ask if music or earplugs are desired to mask environmental noise. | Reduces external stimuli for a highly irritable, hyperactive nervous system. |
| 10 | Administer propranolol or sedatives strictly as ordered. | Reduces the sympathetic nervous system overdrive, allowing the patient to finally rest. Expected Outcome: Patient feels rested upon awakening. |
| 5. Excessive Anxiety related to sympathetic stimulation as evidenced by patient statement. | ||
| 11 | Provide accurate information about the disorder; explain that proper medical treatment *will* correct these terrifying symptoms. | Fear of the unknown produces anxiety. Understanding the physical cause of their emotional lability provides massive relief. |
| 12 | Offer massage, music, or other relaxation techniques; administer prescribed anti-anxiety agents or beta-blockers. | Promotes active physical and mental relaxation. Expected Outcome: Patient experiences and states reduced anxiety. |
| 6. Risk for Injury related to hypermetabolic state and extreme eye involvement (Exophthalmos). | ||
| 13 | Administer lubricating saline eye drops aggressively. Advise the use of dark, tight-fitting glasses. Gently tape eyes shut with non-allergenic tape for sleeping if required. | Protects protruding eyes from severe drying, light damage, and physical injury (since eyelids may fail to fully close over the bulging globe). |
| 14 | Elevate the head of the bed and provide a strict low-sodium diet. | Gravity drainage and low sodium significantly decrease fluid accumulation (edema) behind the eyes. |
| 15 | Teach the patient to report eye pain or vision changes IMMEDIATELY. | These are cardinal signs of pressure crushing the optic nerve, which causes permanent blindness if uncorrected. Expected Outcome: Patient remains safe and free from injury. |
- 7. Fatigue related to hypermetabolic state with massively increased energy requirements.
- 8. Knowledge Deficit related to disease process, prognosis, signs/symptoms, and long-term treatment.
All pre-operative nursing care should be provided identically to routine surgical procedures, with several highly specific additions:
- Achieving Euthyroid State: The patient must be physically euthyroid (normal hormone levels) before the operation. Antithyroid drugs are used to suppress TH secretion, and specific iodine preparations (like Lugol's solution) are given prior to surgery.
Rationale: Iodine profoundly reduces the size and the intense vascularity of the thyroid organ, massively diminishing the chance of catastrophic hemorrhage during the operation. - Nutrition & Cardiac Status: Provide a highly nutritious diet to counteract the ravages of hyperthyroidism. Heavily evaluate cardiac status (ECG) to screen for existing cardiac complications.
- Emotional & Physical Prep: Explain the surgical procedures and approximate duration. Ensure the patient understands they will wake up with neck drain tubes and IV lines.
Post-operative care involves routine surgical management coupled with hyper-vigilant monitoring of the neck and airway.
- Monitor vital signs continuously, watching specifically for tachycardia and hypotension (classic signs indicating internal hemorrhage).
- Monitor the neck dressing for drainage (a moderate amount of serosanguinous drainage is expected, but bright red or excessive blood is an emergency).
- Watch for subtle signs of bleeding: Frequent swallowing or repeated clearing of the throat, rapid visible swelling of the neck, or any signs of respiratory distress (stridor).
- CRITICAL INTERVENTION: Keep a complete tracheostomy set ready at the bedside for the first 48 hours. A rapid hematoma or severe edema can instantly crush the trachea, requiring emergency surgical airway intervention.
- Watch closely for changes in voice quality (extreme hoarseness or inability to speak), which indicate damage to the recurrent laryngeal nerve.
- Monitor for Hypocalcemia (Tetany): Since the parathyroid glands are embedded on the back of the thyroid, they are easily damaged or accidentally removed. Monitor blood calcium levels (if it falls below 7 mg/dL, prepare for immediate IV calcium replacement).
- Assess for clinical signs of hypocalcemic tetany: Irritability, severe twitching, intense spasms of hands and feet, and tingling/numbness of the toes and around the mouth.
- Positive Chvostek's Sign (Weiss sign): When the facial nerve is gently tapped at the angle of the jaw (masseter muscle), the facial muscles on that same side will momentarily contract or twitch due to severe hyperexcitability of the nerves.
- Positive Trousseau's Sign: Inflating a blood pressure cuff above systolic pressure for a few minutes elicits painful carpopedal spasms (claw-like curling of the hand and fingers).
- Keep the patient strictly in a Semi-Fowler's position. This facilitates optimal breathing mechanics and uses gravity to decrease surgical site edema.
- Move the patient exceptionally carefully. Provide adequate, rigid support to the head and neck using sandbags or pillows on either side.
Rationale: To prevent any sudden extension or flexing of the neck, which puts catastrophic tension on the fresh surgical sutures. - Administer pain medications as needed and prescribed.
- Teach the patient exactly how to take their new medications and the absolute importance of taking thyroid replacement medicine regularly (often for the rest of their lives).
- Teach the patient the distinct clinical signs of both returning hyperthyroidism and newly developed hypothyroidism to report to the physician.
- Emergency Warning: Instruct them to immediately report high fever, tachycardia, or extreme irritation/anxiety, as these are late-occurring signs of a post-operative thyroid storm.
- Emphasize the strict necessity of routine, lifelong follow-up laboratory testing (TSH, Free T4) to ensure hormone replacement dosages are perfectly calibrated.
Quick Quiz
Hyperthyroidism Quiz
Medical Nursing - mobile-friendly and focused practice.
Privacy: Your details are used only for quiz tracking and certificates.
Hyperthyroidism Quiz
Medical Nursing
Preparing questions...
Choose your answer and keep your streak alive.
Great effort.
Here is your quick performance summary.