Microbiology: Oncogenic Viruses & Hepatitis C (HCV)
Module Overview
This master guide provides an exhaustive, deeply detailed exploration of oncogenic viruses, with a primary focus on the Hepatitis C Virus (HCV). We will explore the precise molecular mechanisms of viral replication, the complex immunological warfare between the virus and the human host, the pathophysiology of viral-induced cancer, and the advancements in diagnostic and antiviral therapies.
Principles of Tumor Viruses and Oncogenesis
Before diving into the specifics of Hepatitis C, it is absolutely essential to establish a foundational understanding of how microscopic viruses can hijack cellular machinery to cause macroscopic tumors (cancer).
What is Oncogenesis?
Oncogenesis (also referred to interchangeably as carcinogenesis or tumourigenesis) is the complex, multi-step biological process by which normal, healthy, carefully regulated cells are fundamentally transformed into chaotic, rapidly dividing cancer cells.
Pathophysiology: This malignant transformation does not happen overnight. It results from a combination of severe genetic mutations, a state of chronic, unresolved inflammation, or the catastrophic disruption of normal cell cycle regulation and apoptosis (programmed cell suicide). When the brakes (tumor suppressor genes) are removed, and the accelerator (oncogenes) is jammed down, cancer develops.
Key Triggers of Oncogenesis:
- Chemical carcinogens: e.g., Tobacco smoke, asbestos, aflatoxin.
- Radiation: e.g., Ultraviolet (UV) light, X-rays, gamma radiation.
- Inherited Genetic mutations: e.g., BRCA1/BRCA2 mutations in breast cancer.
- Oncogenic Viruses: The focus of this module.
What are Oncogenic Viruses (Tumour Viruses)?
An oncogenic virus is an infectious viral agent that is inherently capable of causing or significantly contributing to the development of cancer within a host organism. These viruses orchestrate cancer through three primary, distinct mechanisms:
- Direct Oncogenesis: The viral DNA physically inserts (integrates) itself directly into the host cell's genome. In doing so, it can inadvertently place highly active viral promoters next to human proto-oncogenes, mutating them into hyperactive oncogenes (genes that drive uncontrolled cell division).
- Indirect Oncogenesis: The virus does not alter the host DNA directly. Instead, it causes decades of chronic inflammation. The constant immune attack destroys tissue, forcing the remaining cells to rapidly regenerate. This endless cycle of tissue destruction and rapid, panicked regeneration dramatically increases the statistical likelihood of spontaneous DNA replication errors.
- Tumor Suppression Sabotage: The virus produces specific malignant proteins that physically seek out, bind to, and deactivate the host's critical tumour suppressor genes (such as the p53 guardian protein or the Retinoblastoma (Rb) protein). Without these cellular "police officers," mutated cells are allowed to divide freely.
Recognized Oncogenic Viruses in Human Medicine:
| Virus |
Associated Malignancy |
Mechanism Type |
| HPV (Human Papillomavirus) |
Cervical, Anal, and Oropharyngeal cancers. |
Direct (E6 and E7 proteins degrade p53 and Rb). |
| HBV (Hepatitis B Virus) |
Liver cancer (Hepatocellular Carcinoma). |
Direct Integration + Indirect Inflammation. |
| EBV (Epstein-Barr Virus) |
Burkitt's Lymphoma, Nasopharyngeal carcinoma, Hodgkin's Lymphoma. |
Direct (Immortalizes B-lymphocytes). |
| HTLV-1 (Human T-lymphotropic virus) |
Adult T-cell leukemia/lymphoma. |
Direct (Tax protein drives T-cell proliferation). |
| HCV (Hepatitis C Virus) |
Liver cancer (Hepatocellular Carcinoma). |
Strictly Indirect (Chronic inflammation/ROS). |
Note: The World Health Organization (WHO) explicitly classifies HCV as a Group 1 Carcinogen (known to cause cancer in humans).
Virology Expansion: Direct vs. Indirect
A crucial distinction for your microbiology exams: Hepatitis B (HBV) is a DNA virus that integrates directly into the host genome, making it a Direct Oncogen. Hepatitis C (HCV) is an RNA virus. Its replication happens entirely in the cytoplasm, and it never integrates into the host DNA. Therefore, HCV causes cancer strictly through Indirect Oncogenesis (driven by decades of chronic inflammation, cirrhosis, oxidative stress, and viral proteins disrupting normal cell cycle checkpoints).
Introduction to Hepatitis C & Epidemiology
Basic Viral Classification
- Full Name: Hepatitis C Virus (HCV).
- Family: Flaviviridae. (To put this into context, this family also includes highly dangerous classical flaviviruses like Yellow Fever, Dengue Virus, Zika Virus, West Nile Virus, and various animal pestiviruses).
- Genus: Hepacivirus.
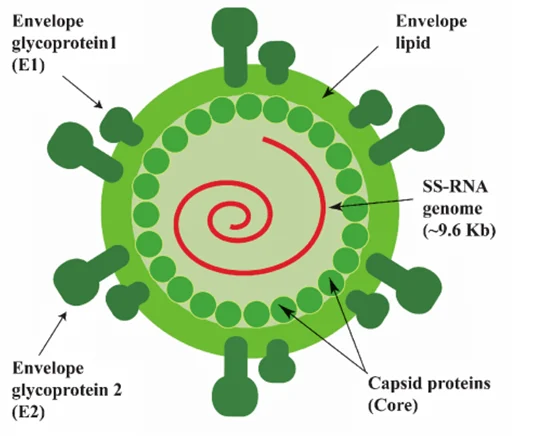
- Viral Architecture: Enveloped, positive-sense single-stranded RNA (+ssRNA) virus.
- History: Discovered relatively recently in 1989. Before its official identification, it was mysteriously referred to in medical literature as "Non-A, Non-B Hepatitis."
Epidemiological Statistics and Global Burden
HCV is an insidious, silent pandemic. It represents a massive global health concern and remains a leading indication for liver transplantation worldwide.
- Historical Exposure: Approximately 170 million people worldwide have been exposed to or infected by the virus at some point in their lives.
- Active Chronic Burden: Currently, an estimated 50 million people are living with chronic, active HCV infections globally.
- High-Burden Geographic Regions: Central and East Asia, North Africa & the Middle East, Sub-Saharan Africa, and Eastern Europe. (Specific Example: Egypt has historically had one of the highest HCV prevalence rates in the world—sometimes exceeding 10-15% of the population—due to historical mass-treatment campaigns for schistosomiasis using unsterilized, shared glass syringes in the mid-20th century).
Historical WHO Prevalence Data (1999 estimates):
- Africa: 5.3%
- Eastern Mediterranean: 4.6%
- Western Pacific: 3.9%
- South-East Asia: 2.15%
- Americas: 1.7%
- Europe: 1.03%
- Total Global Average: 3.1%
Transmission Routes
HCV is transmitted strictly through exposure to infected blood. It is a robust virus that can survive outside the body at room temperature on environmental surfaces for up to 3 weeks.
Primary Routes (Blood-to-blood contact):
- Injection drug use (IVDU): Sharing contaminated needles, syringes, or drug-preparation equipment accounts for a staggering 60% of modern infections.
- Blood Transfusions & Organ Transplants: Accounts for 10% of historical infections. This was the primary mode of transmission before mandatory, highly sensitive nucleic acid blood screening protocols were implemented globally in the early 1990s.
- Occupational Exposure: Needlestick injuries in healthcare workers account for roughly 4% of cases.
- Unregulated Tattoos & Piercings: Using unsterilized ink or needles in unregulated environments carries a significant risk.
Less Common Routes:
- Sexual Transmission: Accounts for ~15% of cases according to CDC historical data. However, the risk is generally considered low in monogamous heterosexual relationships. The risk increases exponentially if there are co-infections (like HIV), multiple partners, or practices that induce mucosal trauma resulting in blood exposure.
- Perinatal / Mother-to-Child (Vertical Transmission): Less common, grouped into the 1% "Other" category along with nosocomial (hospital-acquired) and iatrogenic (medically induced) sources. Roughly 4-6% of infants born to HCV-positive mothers will contract the virus.
- Unknown Sources: Approximately 10% of patients have no identifiable risk factors.
Memory Hack
The Hepatitis Viruses Transmission Routes
To perfectly remember how the major Hepatitis viruses are transmitted, use this simple mnemonic rule:
- Hepatitis A & E = Ate & Eaten: Transmitted via the Fecal-Oral route (contaminated food and water). They cause acute, self-limiting infections.
- Hepatitis B, C, & D = Blood-borne, Contact (Sexual), Drug use: Transmitted via bodily fluids. These are the ones that lead to chronic liver disease and cancer.
Molecular Virology of Hepatitis C
A. Virion Structure
The physical structure of the HCV virion is perfectly adapted for entering human liver cells (hepatocytes).
- Nucleocapsid: The virus features a tightly packed, Icosahedral nucleocapsid that houses the delicate viral RNA.
- Lipid Envelope: This capsid is surrounded by a host-derived lipid envelope. The virus literally steals a piece of the human cell membrane as it exits, using it as a disguise.
- Glycoproteins (E1 and E2): Embedded deep within this stolen lipid envelope are the heavily glycosylated viral envelope proteins E1 and E2. These proteins act as the "keys" that mediate host cell entry by binding to highly specific receptors on the surface of human hepatocytes (such as CD81, Scavenger Receptor B1, Claudin-1, and Occludin).
B. Viral Genome Architecture
The HCV genome is a masterclass in biological efficiency. It consists of a 9.6 kilobase (kb) positive-strand RNA genome (+ssRNA). Because it is "positive-sense," the viral RNA acts exactly like human messenger RNA (mRNA). As soon as it enters the cell, human ribosomes can read it immediately.
- Untranslated Regions (UTRs): These are sequences at the ends of the genome that do not code for proteins but are vital for survival.
- 5' UTR (The Hijacker): Contains an IRES (Internal Ribosome Entry Site). Physiology Expansion: Normal human mRNA requires a special "5' cap" for human ribosomes to recognize it and bind to it. The HCV IRES is a complex, 3D folded RNA structure that physically "hijacks" the human ribosome, forcing it to lock on and translate the viral RNA without needing a standard 5' cap!
- 3' UTR: A highly structured noncoding region absolutely essential for the initiation of RNA replication.
- Open Reading Frame (ORF): The vast majority of the genome is one continuous reading frame. It encodes a single, massive "polyprotein" consisting of approximately 3,000 amino acids.
C. The Polyprotein Cleavage Products (Crucial Exam Material)
A giant 3,000-amino-acid string is useless. It must be chopped up. This polyprotein is systematically cleaved by both host cellular proteases and the virus's own viral proteases into 10 distinct structural and non-structural (NS) proteins.
| Protein |
Function / Characteristics |
| C (Core) |
Forms the physical viral nucleocapsid. Importantly, it is a primary weapon for immune evasion, actively targeting and blocking the host's JAK-STAT signaling pathway (preventing interferon response). |
| E1 & E2 |
The Envelope glycoproteins. E2 contains HVR-1 and HVR-2 (Hypervariable Regions). These regions mutate wildly and constantly, allowing the virus to rapidly change its surface "face" and completely evade neutralizing antibodies. E2 also actively targets and suppresses the host enzyme PKR. |
| p7 |
Thought to function as a viroporin—a tiny viral ion channel that punches holes in membranes, essential for efficient virus assembly and release from the cell. |
| NS2 |
A Zinc-dependent proteinase. Its only job is to cleave the junction between itself and NS3. |
| NS3 |
A multi-functional powerhouse and central therapeutic target. It acts as a Zinc-dependent proteinase, a serine protease, and an RNA helicase (unwinding RNA). (This is a major target for modern antiviral drugs!). |
| NS4A |
Acts as a vital, stabilizing cofactor for the NS3 protease, anchoring it to intracellular membranes. |
| NS4B |
Induces the massive rearrangement of the host's Endoplasmic Reticulum (ER), forcing the creation of a complex "membranous web" inside the host cell. This creates a hidden, secure factory strictly for viral replication, shielded from cellular immune sensors. |
| NS5A |
A heavily phosphorylated zinc-metalloprotein. Its exact function is heavily researched; it is an essential structural component of the replicase complex and heavily modulates host immune responses (specifically targeting and inhibiting PKR). |
| NS5B |
The RNA-dependent RNA polymerase (RdRp). This is the enzyme that physically copies the viral RNA to make new viral genomes. |
The Discovery of Protein F (Frameshift Protein):
Advanced research has revealed a newly discovered protein produced by a ribosomal "frameshift" mutation (where the ribosome slips and reads the code out of phase) around codon 11 of the Core protein region. Its exact function remains a mystery, but we know it is produced during active infection because infected individuals reliably produce antibodies against it.
Genotypes & The Quasispecies Concept
Genotypes
HCV is extremely genetically diverse. It is divided into 6 major genotypes (1 through 6), which are distributed differently worldwide. Furthermore, these genotypes are broken down into numerous subtypes (e.g., 1a, 1b, 2a) and individual isolates based on nucleotide diversity. For example, Genotype 1 is the most common in the United States and Europe, while Genotype 4 dominates in the Middle East and Egypt.
The Quasispecies Concept
HCV possesses an astronomically high mutation rate. Why? Because its polymerase enzyme (NS5B) completely lacks "proofreading" ability (exonuclease activity). When a human cell copies DNA, it checks for typos and fixes them. When NS5B copies HCV RNA, it makes millions of random typos and leaves them there.
As a direct result, a single infected patient does not just carry one uniform virus. Instead, their blood contains a massive, swirling, complex swarm of millions of closely related, yet distinct mutant viruses. This swarm is called a Quasispecies.
Evolutionary Biology Application: This swarm functions as a unified evolutionary unit of selection. It brilliantly balances rapid mutation with high adaptability. If you give a patient a drug, or if their immune system generates a new antibody, 99% of the viral swarm might die. But because the swarm is so diverse, there is almost certainly a mutant in the remaining 1% that is perfectly immune to the drug or antibody. That mutant survives, replicates, and repopulates the liver.
Applied Clinical Question: Research Challenges
Case: A pharmaceutical company is struggling to develop new treatments and vaccines for Hepatitis C. According to clinical history, what were the major research roadblocks associated with studying HCV in the laboratory?
Answer: Historically, HCV research was severely hindered by three massive roadblocks:
- There was no reliable cell culture system (for decades, the virus outright refused to grow in laboratory petri dishes).
- There was no small animal model (HCV only naturally infects humans and chimpanzees, making ethical and affordable testing nearly impossible).
- The Quasispecies phenomenon. Its ability to rapidly mutate its Hypervariable Regions (HVR) makes finding stable, unchanging viral targets for vaccine development exceedingly difficult.
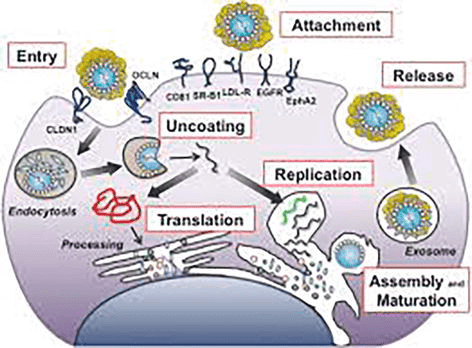
D. The Complete HCV Lifecycle (Cellular Infection Process)
To fully understand how Hepatitis C replicates and how direct-acting antivirals (DAAs) stop it, we must trace the exact chronological pathway the virus takes from the moment it encounters a human hepatocyte (liver cell) to the moment it bursts forth as a new, mature virion. The HCV lifecycle occurs entirely within the cell's cytoplasm and heavily relies on hijacking the host's lipid (fat) metabolism pathways.
Step 1
Attachment
The virus does not simply bump into a cell and enter; it requires a highly complex, multi-receptor "handshake" to ensure it is invading the correct target (a liver cell). The viral envelope glycoproteins (E1 and E2) bind to an array of specific host surface receptors and attachment factors.
Key Receptors Identified:
- CD81: A critical tetraspanin cell-surface protein.
- SR-B1 (Scavenger Receptor B1): A receptor normally used by the liver to uptake high-density lipoproteins (HDL). The virus disguises itself in lipids to bind here.
- LDL-R: Low-Density Lipoprotein Receptor.
- EGFR & EphA2: Epidermal Growth Factor Receptor and Ephrin receptor A2 act as essential co-factors that signal the cell membrane to prepare for entry.
Step 2
Entry (Endocytosis)
After initial attachment, the virus migrates along the cell surface to the tight junctions between hepatocytes. Here, it interacts with two more crucial tight-junction proteins: CLDN1 (Claudin-1) and OCLN (Occludin). Once this final lock is turned, the cell membrane invaginates (folds inward) and swallows the virus in a process called Clathrin-mediated Endocytosis, trapping the virus inside an intracellular bubble called an endosome.
Step 3
Uncoating
Inside the host cell, the endosome naturally becomes acidic (a drop in pH). The virus uses this acid as a trigger. The acidic environment causes a massive shape change in the viral envelope glycoproteins, forcing the viral lipid envelope to fuse with the endosomal membrane. The virus is "uncoated," and its naked positive-sense RNA genome (+ssRNA) is violently ejected directly into the host cell's cytoplasm.
Step 4 & 5
Translation and Processing
Because the viral RNA is "positive-sense," the human ribosomes immediately mistake it for human mRNA. The viral 5' IRES (Internal Ribosome Entry Site) hijacks the ribosomes located on the Rough Endoplasmic Reticulum (ER).
Translation: The ribosome translates the entire genome into one massive, continuous polyprotein.
Processing: This giant protein thread weaves in and out of the ER membrane. It is rapidly chopped into 10 active pieces (Core, E1, E2, p7, NS2, NS3, NS4A, NS4B, NS5A, NS5B) by both host signal peptidases and the viral NS3/4A protease.
Step 6
Replication
The viral protein NS4B causes the host's ER to physically warp and fold into a complex "membranous web." Inside this shielded web, the viral polymerase (NS5B) goes to work. First, it reads the original +ssRNA and creates a "negative-sense" intermediate template (the green strand). Then, it uses that negative template to continuously print millions of new, identical +ssRNA genomes (the black strands) to be packaged into new viruses.
Step 7 & 8
Assembly, Maturation, and Release
Assembly: HCV assembly is uniquely tied to the liver's fat metabolism. The newly minted viral RNA and the Core proteins gather around host Lipid Droplets located near the ER. The Core protein encases the RNA, forming the nucleocapsid.
Maturation: The capsid pushes into the ER lumen, wrapping itself in a piece of the ER membrane embedded with E1/E2 glycoproteins, forming a mature, enveloped virion.
Release (Exosome Pathway): The mature virus hitches a ride on the cell's Very-Low-Density Lipoprotein (VLDL) secretory pathway. It is transported to the cell surface inside an exosome (a secretory vesicle) and is expelled from the hepatocyte via exocytosis, ready to infect a neighboring cell or enter the bloodstream.
Clinical Presentation & Patterns of Viremia
HCV infection is notoriously stealthy. The clinical progression of the disease is broadly divided into an acute phase and a chronic phase.
1. Acute Phase Infection:
- Asymptomatic Nature: The vast majority of patients are entirely asymptomatic and remain blissfully unaware they have been infected.
- Symptomatic Cases: If symptoms do manifest (usually 2 to 12 weeks post-exposure), they are vague and mild: severe fatigue, nausea, abdominal discomfort, dark urine, and rarely, overt jaundice (yellowing of the skin and sclera due to bilirubin buildup from liver inflammation).
- Spontaneous Clearance: Approximately 15% to 25% of individuals possess an immune system robust enough to successfully clear the virus on their own during this acute phase without ever requiring medical intervention.
2. Chronic Phase (The "Silent" Disease):
- High Chronicity Rate: The vast majority of infected individuals—75% to 85%—fail to clear the virus, leading to a lifelong persistent HCV infection.
- The Silent Progression: Patients can remain completely asymptomatic for 20 to 30 years while the virus slowly and methodically damages the liver. (Note: The liver itself contains no pain receptors; only the outer capsule does. Therefore, a liver can be 90% destroyed by fibrosis without the patient feeling any physical pain in the organ.)
- Disease Trajectory: Chronic Hepatitis → Progressive liver scarring (Fibrosis) → Cirrhosis (irreversible architectural distortion of the liver) → End-stage liver failure or Hepatocellular Carcinoma (HCC).
- Extraintestinal Manifestations: The virus doesn't just damage the liver. The persistent, massive immune response can trigger circulating immune complexes that deposit in other organs, causing severe autoimmune diseases such as Mixed Cryoglobulinemia (proteins clumping in cold blood leading to vasculitis), Glomerulonephritis (kidney damage), Porphyria Cutanea Tarda (skin blistering in sunlight), and Sjögren's syndrome.
3. Patterns of Viremia (Virus levels in the blood):
Monitoring the viral load in a patient's blood over time reveals three distinct patterns that determine their ultimate clinical outcome:
- Drop after peak: The viral load spikes, but then plummets to zero. Indicates successful, robust immune control and permanent viral clearance.
- Drop followed by rebound: The immune system initially mounts a strong defense, dropping the viral load. However, the viral Quasispecies rapidly mutates, evades the attack, and rebounds to high levels, establishing chronic infection.
- Consistent, unrelenting HCV levels: The immune system completely fails to mount an effective initial response, leading to immediate, rapid chronic infection and high, sustained viremia.
The Innate Immune Response & Viral Evasion Strategies
The innate immune system is the body's rapid-response first line of defense. It responds instantly (within hours to 2 days of infection), regardless of what the final outcome of the infection will be.
The Cellular Defense Mechanism (How the cell fights back):
As the HCV RNA genome replicates inside the cytoplasm, it must form dsRNA (double-stranded RNA) intermediates. dsRNA is highly unnatural in a human cell's cytoplasm. To the cell, dsRNA acts as a massive, blaring biochemical alarm bell!
- PKR Activation: The presence of this dsRNA activates an enzyme called Protein Kinase R (PKR). (Physiology Expansion: PKR is a cellular suicide switch. When activated, it phosphorylates translation initiation factors, effectively shutting down ALL protein translation in the cell to starve the virus of manufacturing parts).
- IRF Phosphorylation: Intracellular sensors (like RIG-I and MDA5) detect the viral RNA and trigger a cascade that phosphorylates Interferon Regulatory Factors (IRFs), specifically IRF-3.
- Gene Activation: These phosphorylated IRFs act as transcription factors. They travel directly into the cell's nucleus to upregulate and turn on massive arrays of antiviral gene products (specifically Type I Interferons and interferon-stimulated genes).
- Result: These gene products degrade the viral RNA, trigger apoptosis in infected cells, and warn neighboring cells to raise their shields.
HCV Viral Resistance & Targeting (How the virus wins):
HCV is incredibly successful at causing lifelong chronic infection because its viral proteins are exquisitely evolved to directly target, sabotage, and dismantle the human innate immune pathways!
- NS5A and E2 target PKR: These viral proteins physically bind to and completely inhibit Protein Kinase R. The cell is unable to shut down protein translation, allowing the virus to continuously print new viral parts.
- Core protein targets the JAK-STAT pathway: (Expansion: When Interferon is released, it binds to neighboring cells and activates the JAK-STAT pathway to arm them against incoming viruses). The HCV Core protein chemically blocks this exact pathway, making all neighboring cells "deaf" to the interferon alarm system.
- NS3/4A targets phosphorylated IRF-3: The viral protease NS3/4A acts as molecular scissors. It physically chops up the essential cellular adaptor proteins (like MAVS and TRIF) needed to activate IRF-3. This completely, irreparably shuts down the cell's ability to produce Type I Interferons!
High-Yield Concept
The Protease Weapon (NS3/4A)
The viral protein NS3/4A is a highly specialized protease. Not only does it cut the giant viral polyprotein into usable, individual pieces to build new viruses, but it literally snips the host cell's immune alarm wires (MAVS and TRIF proteins). By cutting these wires, the infected cell cannot alert the rest of the immune system. This dual-action sabotage is exactly why pharmaceutical NS3/4A protease inhibitors are one of the most powerful cures we have today—they stop viral assembly AND restore the cell's ability to call for help!
Adaptive Immune Response & Chronic Dysregulation
If the innate system fails (which it usually does with HCV), the adaptive immune system (T-cells, B-cells) steps in. The speed and vigor of this secondary response dictate whether the patient lives a virus-free life or suffers decades of chronic liver disease.
Characteristics of Individuals Who Successfully Control the Virus:
- IFN-γ (Interferon-gamma): Preferentially and strongly expressed in the liver of recovering patients.
- IFN-γ strongly induces the expression of genes encoding chemokines that attract armies of T-cells right into the inflamed liver tissues.
- It also highly induces proteins associated with antigen processing and presentation (upregulating MHC Class I and II molecules on liver cells so they can "show" the immune system the hidden virus).
- These patients mount highly vigorous, sustained, and multi-specific CD8+ (Cytotoxic) and CD4+ (Helper) T cell responses.
Why Chronic Infections Occur (Adaptive Failure):
The majority of patients develop chronic disease due to two primary failures:
- The patient's immune system is simply unable to mount a broad, HCV-specific T cell response from the beginning.
- There is a strong initial response resulting in apparent viral RNA clearance, but this is followed prematurely by a massive contraction (die-off or exhaustion) of the CD8+/CD4+ cells. With the army depleted, the surviving mutated Quasispecies virus mounts a massive rebound in viremia.
The State of Chronic HCV Immune Dysregulation:
- CD8+ T-cells (Cytotoxic Killers): They experience abnormally low frequencies in the blood and a heavily reduced capacity to kill infected cells. This is a clinically recognized state known as T-cell exhaustion (often driven by the continuous upregulation of inhibitory receptors like PD-1).
- CD4+ T-cells (Helpers): Suffer from severely reduced Interleukin-2 (IL-2) production and demonstrate poor proliferation. Without Helpers, the Killers fail.
- Dendritic Cells (Antigen Presenters): They do not mature normally and exhibit severely impaired stimulatory activity. They fail to effectively capture the virus and "show" it to the T-cells to initiate an attack.
- Natural Killer (NK) & NKT Cells: There is a severe impairment of NK cell cytotoxic (cell-killing) activity and a decreased frequency of NKT cells in the liver. (Note: Clinical trials show this profound impairment is actually fully reversible in patients who respond well to exogenous IFN-α drug therapy!)
The Role of Antibodies (B-Cell Response):
- The role of humoral (antibody) immunity in HCV is surprisingly unclear, contradictory, and historically poorly studied.
- Fascinatingly, the virus can actually be cleared completely by some patients in the absolute absence of detectable antibody responses (proving that CD8+ T-cells are the true heroes of viral clearance).
- The body does produce neutralizing antibodies that attempt to target the E2 envelope glycoprotein. However, as noted earlier, E2 contains Hypervariable Regions (HVR1/HVR2). E2 mutates so astronomically fast that by the time the B-cell manufactures a perfect antibody, the virus has already changed its molecular "face" to evade it. The antibodies are always one step behind the Quasispecies swarm.
The Liver Environment & Hepatocellular Carcinoma (Oncogenesis)
The ultimate tragedy of long-term HCV infection is the development of Hepatocellular Carcinoma (HCC). To understand why this happens, we must look closely at the unique immune environment of the liver.
A. The Normal vs. Infected Liver Environment
The Normal Liver:
- Maintains an intentionally "Immuno-silent" state. (Physiology Expansion: Because the liver receives all nutrient-rich blood straight from the intestines via the portal vein, it is constantly bombarded by harmless food antigens, plant proteins, and dead gut bacteria pieces. If the liver's immune system reacted to all of these foreign bodies, you would be in a constant, fatal state of anaphylactic shock!)
- To maintain this vital silence, activated CD8+ T cells that wander into the normal liver are often trapped and forcibly pushed into apoptosis (programmed cell death) by local hepatic cells to prevent unnecessary immune reactions.
The HCV-Infected Liver:
- The viral invasion breaks the silence. The stressed liver produces Type I IFN and releases chemokines that promote the rapid infiltration of NK cells.
- This induces massive IFN-γ production in the NK cells and expresses chemokines that recruit tens of thousands of highly activated T-cells to the liver matrix.
- Clinical Note: Experimental depletion of NK cells prior to a hepatotropic viral infection leads to the absolute inhibition of the virus-specific T-cell response, resulting in unabated viral replication and severe liver injury.
B. Immune-Mediated Liver Injury & "Bystander Killing"
The mechanisms responsible for liver injury were initially poorly understood. We now know that the Host immune response, NOT the direct viral replication itself, is what physically destroys the liver.
- HCV is actually quite sparse; it typically infects only 1% to 10% of the total hepatocytes in the liver at any given time.
- However, the massive influx of highly active, frustrated CD8+ T-cells into the liver results in the release of a fiery storm of toxic cytokines (like IFN-γ and TNF-α) and deadly granules (perforin and granzyme). These toxic compounds are indiscriminately sprayed into the tissue, destroying vast numbers of uninfected, innocent hepatocytes in a tragic, highly destructive process known as "Bystander Killing".
C. The Road to Hepatocellular Carcinoma (Indirect Oncogenesis)
Because HCV does NOT integrate into host DNA (unlike HBV), its mechanism of causing cancer is entirely indirect, stemming from the chronicity of the battle.
- The ongoing "Bystander Killing" by the immune system causes massive, unrelenting hepatocyte death and chronic Inflammation.
- To survive the destruction, the liver goes into overdrive, constantly attempting to regenerate. This extremely high turnover rate in hepatocytes pushes cells to divide faster than their DNA repair mechanisms can handle, leading to numerous erroneous genome replications.
- Furthermore, the chronic inflammation involves millions of immune cells generating massive amounts of Oxidative stress and Reactive Oxygen Species (ROS), which bathe the liver cells and physically shatter cellular DNA strands.
- This endless cycle of tissue damage → ROS generation → rapid cellular repair strongly activates Hepatic Stellate Cells. These cells lay down massive amounts of collagen, causing widespread Fibrosis (scarring) and eventually Cirrhosis (a hard, nodular, failing liver).
- The cirrhotic liver, full of rapidly dividing cells containing ROS-damaged DNA, creates a highly unstable "pre-cancerous niche" that ultimately promotes the transformation of these cells into Hepatocellular Carcinoma (HCC).
- Additional Factor: Viral proteins (like NS5A and Core) remaining in the few infected cells further disrupt normal cell cycle regulation and prevent apoptosis, sealing the malignant fate of the tissue.
Summary Analogy
HCV Oncogenesis: The Warzone
HCV does NOT directly mutate your DNA. Instead, think of the liver as a city and the infection as a Warzone:
- HCV hides in a few buildings (cells).
- The immune system (CD8+ cells) drops massive "bombs" (TNF-α, ROS) to kill the virus, blowing up thousands of innocent, healthy buildings alongside the infected ones (Bystander killing).
- The liver city tries to rebuild the destroyed buildings as fast as possible (High cell turnover/regeneration).
- Rebuilding quickly in a toxic, oxidative warzone causes the construction workers to make terrible mistakes (DNA replication errors).
- Over decades, the mistakes pile up, and the city becomes filled with chaotic, uncontrolled, collapsing structures. This is Hepatocellular Carcinoma.
Diagnostics, Vaccine Status, and Antiviral Therapeutics
IX. The Vaccine Challenge: Why is there no HCV Vaccine?
Unlike Hepatitis B (HBV) and Human Papillomavirus (HPV), for which we possess highly effective, globally distributed routine vaccines, there is currently NO approved vaccine for Hepatitis C.
The Biological Roadblocks preventing vaccine development include:
- Astronomical Genetic Variability: There are 6 major genotypes with dozens of highly distinct subtypes. A vaccine formulated to recognize Genotype 1 will likely offer zero protection against Genotype 4.
- The Quasispecies Moving Target: As discussed, the lack of proofreading by the NS5B polymerase creates a rapidly shifting swarm of viruses. The virus mutates its surface envelope proteins faster than the immune system—or a vaccine-induced antibody response—can lock onto them.
- Active Immune Evasion: The viral proteins (NS5A, E2, Core) actively suppress and disarm the host's innate and adaptive immune responses, making it hard for a vaccine to trigger strong memory cells.
- Lack of Animal Models: Since HCV only naturally infects humans and chimpanzees (and testing on chimpanzees is highly restricted/banned in most of the world), testing new, experimental vaccine candidates is incredibly difficult, expensive, and ethically complex.
X. Investigations & Clinical Diagnostics
Accurately diagnosing HCV requires a strict two-step process. You cannot rely on a single blood test to determine if a patient is currently harboring an active infection.
Step 1: Initial Screening (Indirect Serological Test)
- The Test: HCV Antibody (anti-HCV) utilizing Enzyme-Linked Immunosorbent Assay (ELISA) or a Rapid Diagnostic Test (RDT).
- Positive Result Meaning: It ONLY indicates that the patient was exposed to HCV at some point in their life and their immune system created antibodies. It does not mean they are currently sick. (Remember, 25% of people clear the virus naturally, but their antibody test will remain positive for life!).
- Negative Result Meaning: Generally rules out infection entirely.
- Exception 1 (The Window Period): It takes the human body 8 to 12 weeks after initial exposure to produce enough antibodies to be detected by a lab test. If a nurse suffers a contaminated needlestick injury yesterday, the antibody test today will be negative, even if the virus is currently ravaging their liver.
- Exception 2 (Immunocompromised State): Patients with advanced, untreated HIV/AIDS or those on heavy immunosuppressants may fail to produce antibodies entirely, yielding a false-negative result.
Step 2: Confirmatory Testing (Direct Virological Tests)
If the screening antibody test is positive, the clinician MUST perform a "direct" test to look for the physical presence of the virus itself.
- HCV RNA (Nucleic Acid PCR Testing): The absolute Gold Standard for diagnosis.
- Qualitative RNA: A highly sensitive "Yes/No" test. It simply tells you if viral RNA is present in the blood.
- Quantitative RNA (Viral Load): Measures the exact amount of viral copies per milliliter of blood. This is absolutely essential for establishing a baseline before treatment and monitoring therapeutic success.
- HCV Core Antigen (HCVcAg): A test that detects the physical Core viral protein rather than the RNA. It is utilized heavily in resource-limited settings as a significantly cheaper, more accessible alternative to complex PCR machines; it correlates excellently with active, high viral loads.
Applied Clinical Question: Pediatric Testing
Case: A baby is born to an HCV-positive mother. The enthusiastic intern wants to run an HCV Antibody (ELISA) test on the newborn at 1 month of age to see if transmission occurred. Why is this a profound medical error, and what should be done instead?
Answer: For all children under 18 months of age, HCV antibody tests are completely unreliable. This is because the newborn passively carries the mother's maternal IgG antibodies, which crossed the placenta during the third trimester. The antibody test would be overwhelmingly positive even if the baby is perfectly healthy and virus-free. Instead, an HCV RNA PCR is the strictly preferred test to detect actual viral presence, typically performed when the infant is at least 2 months old to allow viral replication to reach detectable thresholds.
XI. Evolution of HCV Therapy
The treatment landscape for Hepatitis C has undergone a total, miraculous revolution over the last decade, transitioning from highly toxic, poorly effective "hit-or-miss" injections to nearly 100% effective, easily tolerated oral pills.
A. Historical Therapy (Standard/Current in older notes):
- The Combination Regimen: Weekly injections of Pegylated Interferon-alpha (PEG-IFN) combined with daily oral doses of Ribavirin (a broad-spectrum nucleoside analog).
- Mechanism of Action: Massive, systemic suppression of protein synthesis, enhancement of cellular apoptosis, and catastrophic degradation of the viral plus-strand RNA via induced mutagenesis.
- Efficacy: Disappointing. Only 50% to 80% effective, heavily dependent on the specific viral genotype (Genotype 1 was notoriously resistant).
- The "Nightmare" Side Effects: The treatment was often described by patients as worse than the disease itself.
- Flu-like symptoms: Extreme, debilitating tiredness, high fevers, and severe myalgia after every injection.
- Psychiatric: Profound trouble with concentration, severe mood instability, and the induction of deep clinical depression (frequently leading to suicidal ideation).
- Hematologic (Blood) Toxicity: Severe bone marrow suppression leading to dangerous neutropenia (low white cells) and thrombocytopenia (low platelets).
- Ribavirin Toxicity: Directly and fiercely toxic to red blood cells, causing severe Hemolytic Anemia in a large percentage of patients.
B. The Revolution: Direct-Acting Antivirals (DAAs):
Introduced to the global market in the mid-2010s, DAAs fundamentally changed HCV from an agonizing, chronic life sentence into a rapidly curable disease.
- Cure Rate: Exceeds a phenomenal 95% to 99% across all known genotypes.
- Treatment Course: Remarkably short duration, requiring only 8 to 12 weeks of strictly oral, once-daily pills, with near-zero side effects compared to Interferon.
- Standard Global Regimens:
- Sofosbuvir / Velpatasvir: Highly favored because it is suitable for ALL genotypes (1 through 6). It is considered "Pan-genotypic."
- Glecaprevir / Pibrentasvir: Recommended for 8 or 12 weeks depending strictly on the presence or absence of advanced cirrhosis.
- Sofosbuvir / Daclatasvir: Highly common and incredibly cost-effective in resource-limited and developing settings.
Memory Hack
DAA Naming Suffixes
DAAs are precision-engineered to target specific non-structural viral proteins. You can perfectly identify a drug's exact molecular target simply by looking at the suffix of its generic name:
- -previr (e.g., Telaprevir, Glecaprevir): Targets the NS3/4A Protease (P for Protease).
- -asvir (e.g., Daclatasvir, Velpatasvir): Targets the NS5A complex (A for 5A).
- -buvir (e.g., Sofosbuvir): Targets the NS5B RNA-dependent RNA Polymerase (B for 5B). (Mechanism detail: Sofosbuvir acts as a defective nucleotide. When the NS5B polymerase tries to insert it into the growing RNA chain, it acts as a "chain terminator," permanently halting viral replication).
XII. Novel and Experimental Drug Therapies
While DAAs are highly successful, medical research continues deeply into allosteric inhibitors, host-targeting agents, and molecular "warheads" to ensure treatment options exist for the rare patients who develop DAA resistance or suffer from end-stage liver failure.
- Non-nucleoside inhibitors (NNIs): These molecules target the RdRp (NS5B polymerase) at distinct, separate binding sites away from the active center. They work via allosteric inhibition (they bind to the side of the enzyme, physically changing the 3D shape of the enzyme so it can no longer function). Examples include Benzothiadiazine and Benzimidazole chemical derivatives.
- Protease Inhibitors (BILN 2061): A highly specific peptidomimetic compound that physically blocks the NS3 active site. Early clinical trials showed a massive, rapid decline in viral load within 48 hours, though some patients experienced rebounds as the Quasispecies mutated.
- Cyclosporin A (CsA): Traditionally an immunosuppressive drug used for transplant patients, CsA was discovered to strongly bind to cyclophilins (critical human host proteins that the HCV virus absolutely needs to fold its own proteins properly). By blocking the host's calcineurin pathway, it inhibits the activation of genes essential for T-cell activation. (Crucial Note: The highly related immunosuppressive drug FK506 does NOT suppress HCV replication, making Cyclosporin A's specific antiviral mechanism unique and fascinating).
- Arsenic Trioxide: Historically a poison, modern research shows it surprisingly and potently inhibits HCV replication at submicromolar (extremely low) concentrations without displaying significant toxicity to the human host cells.
RNA-Based Genetic Treatments:
The bleeding edge of antiviral research involves manipulating RNA directly.
- RNA Interference (RNAi): A technique that utilizes the cell's own natural defense machinery (the Dicer enzyme and the RISC complex) to seek out, recognize, and physically chop up specific viral RNA sequences. It is highly sequence-specific and lethal to the virus.
- Small RNAs (Decoys): These engineered molecules act as biochemical "decoys." By overexpressing artificial viral RNA elements in the cell, they act like a sponge, binding up all the viral regulatory proteins and preventing them from binding to the real viral RNA. This effectively starves the virus of its machinery and halts gene expression.
- siRNAs (Small Interfering RNAs): These are designed to target and silence the human cellular cofactors that HCV desperately needs to survive (such as the proteins La, PTB, and hVAP-33). If the virus cannot find its required "human helpers," it cannot replicate, regardless of how aggressively it mutates.
XIII. Conclusion & Prevention Strategies
Summary: Hepatitis C Virus remains a stealthy, highly adaptable oncogenic virus and a major global health concern. However, the advent of modern Direct-Acting Antivirals (DAAs) offers a realistic, achievable hope for the global elimination of the disease. Curing HCV is paramount because it doesn't merely clear a viral infection—it significantly and permanently halts the ongoing liver inflammation, thereby massively reducing the long-term risk of the patient developing fatal Hepatocellular Carcinoma.
Prevention Strategies (Since no vaccine exists):
- Implementation of highly sensitive, routine nucleic acid blood screening protocols before all transfusions and organ transplants.
- Aggressive public health Harm Reduction programs (such as sterile needle and syringe exchange programs for IV drug users).
- Strict adherence to Universal Health Precautions (safe needle disposal, double-gloving, and proper sterilization of medical/dental/tattoo equipment in all clinical settings).
List of References
- World Health Organization (WHO). (1999). Global surveillance and control of hepatitis C. Report of a WHO Consultation organized in collaboration with the Viral Hepatitis Prevention Board, Antwerp, Belgium.
- Centers for Disease Control and Prevention (CDC). (2020). Hepatitis C Questions and Answers for Health Professionals. Atlanta, GA: US Department of Health and Human Services.
- Moradpour, D., & Blum, H. E. (2004). A primer on the molecular virology of hepatitis C. European Journal of Gastroenterology & Hepatology, 16(11), 1297-1301.
- Ahmad, A., et al. (2004). Natural killer cells and hepatitis C virus infection. Immunology, 112(1), 7-16.
- Lindenbach, B. D., & Rice, C. M. (2005). Unravelling hepatitis C virus replication from cellular to molecular levels. Nature Reviews Microbiology, 3(8), 596-606.
- Pawlotsky, J. M. (2014). New hepatitis C therapies: the toolbox, strategies, and challenges. Gastroenterology, 146(5), 1176-1192.
- Guidotti, L. G., & Chisari, F. V. (2006). Immunobiology and pathogenesis of viral hepatitis. Annual Review of Pathology: Mechanisms of Disease, 1(1), 23-61.