Infection Mitigation Measures
Introduction to Infection Mitigation Measures
Infection mitigation measures in healthcare refer to the specific, rigorously applied actions taken to prevent and control the spread of infections within healthcare settings. This comprehensive guide details the foundational protocols, hierarchies of safety, and transmission-based precautions required to keep both patients and providers safe.
I. Introduction & Definition of Terms
Primary Goals of Infection Control:
- To remove or mitigate infection risks: Proactively identifying hazards before they cause harm.
- To completely stop the "Chain of Transmission": Pathogens require a source, a mode of travel, and a susceptible host. Infection control aims to sever this chain at the most vulnerable link.
Clinical Importance: Hospital-Acquired Infections (HAIs)
Infection mitigation measures are of paramount importance in preventing and reducing the risk of Hospital-Acquired Infections (HAIs), historically known as nosocomial infections.
HAIs are infections that a patient contracts while receiving care for another condition (strictly defined as an infection appearing 48 hours or more after hospital admission, or within 30 days of discharge). Hospitals are high-risk environments because they house highly vulnerable, immunocompromised patients and utilize invasive devices that bypass the body's natural anatomical barriers.
The "Big Four" Common HAIs:
- MRSA (Methicillin-resistant Staphylococcus aureus): A highly antibiotic-resistant superbug often spread via contaminated hands of healthcare workers or shared equipment (like stethoscopes).
- C. diff (Clostridioides difficile): A spore-forming bacterium causing severe, life-threatening diarrhea, usually triggered after normal gut flora is wiped out by broad-spectrum antibiotics.
- CLABSI (Central Line-Associated Bloodstream Infections): Deadly infections occurring when bacteria travel down an intravenous central line directly into the heart/bloodstream.
- CAUTI (Catheter-Associated Urinary Tract Infections): The most common HAI, occurring when urinary catheters act as a physical highway for bacteria to ascend into the bladder.
Dual Protection Principle
The implementation of infection control measures is never one-sided. It is a mutually protective framework crucial to ensure the safety of both the highly vulnerable patients and the frontline Healthcare Personnel (HCP) operating within the facility.
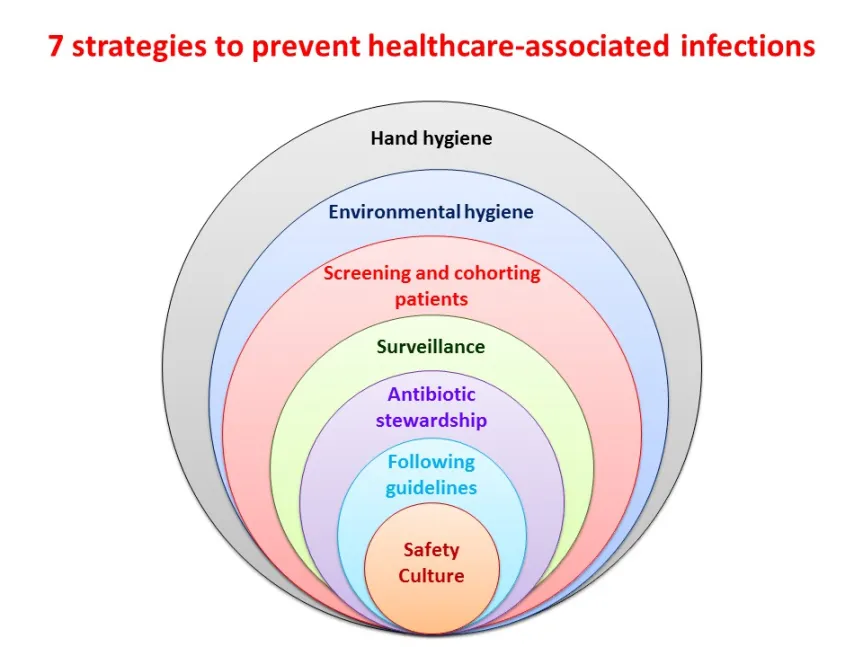
II. The Hierarchy of Controls
The Hierarchy of Controls is a step-by-step framework utilized by occupational health bodies (like NIOSH and the CDC) to eliminate or reduce hazards. It outlines a systematic approach to managing infectious agents.
Why a "Hierarchy"?
The hierarchy is strictly ordered from Most Effective to Least Effective. The premise is logical: controlling the hazard structurally at the source is vastly superior to relying on human memory, behavior, and compliance at the very end of the chain.
- Benefits of the Hierarchy: Improved overall safety, increased productivity (fewer sick days for staff, shorter hospital stays for patients), strict regulatory compliance, and a proactive (rather than reactive) safety culture.
The Five Levels of Control
- Elimination (Physically remove the hazard)
This is the most effective measure because it completely removes the risk of exposure from the environment.
- Laboratory Application: Discontinuing the use of a highly virulent pathogen strain in a teaching lab and replacing it with a completely harmless, non-infectious organism.
- Clinical Application: If a patient has a highly contagious disease (e.g., active COVID-19) but needs a non-urgent elective surgery, you delay the surgery until they are no longer infectious.
- Staff Protocol: Enforcing strict "stay home if sick" policies. An infected nurse cannot spread the flu to the ICU if they are physically eliminated from the ICU.
- Substitution (Replace the hazard)
When a source of infection cannot be entirely eliminated, substitutions should be implemented to reduce the risk to a more manageable level.
- Laboratory Application: Using alternative, less hazardous reagents, or using a less virulent, attenuated vaccine-strain of a virus for research instead of the wild-type virus.
- Clinical Application: Utilizing Virtual Consultations (Telemedicine) via phone or video instead of in-person visits for routine checkups during a viral outbreak.
- Requirement: Always compare the potential new risks of the substitute to the original risks to ensure a net safety gain.
- Engineering Controls (Isolate people from the hazard)
These controls are physical, structural changes built into the facility. They reduce the risk of exposure directly at the source, without relying on human memory or behavior to act.
- Airborne Infection Isolation Rooms (AIIRs): Specially designed hospital rooms with negative air pressure that physically trap airborne pathogens and vent them safely outside.
- Biosafety Cabinets: Used in laboratories; they utilize laminar flow and HEPA filters to contain dangerous aerosols while a microbiologist works.
- Automated Systems: Installing hands-free sinks, automated soap dispensers, and motion-sensor door openers so contaminated hands never touch physical surfaces.
- Administrative Controls (Change the way people work)
These rely on facility policies, written procedures, training, and human adherence to the rules. Because humans make mistakes, this is less effective than engineering.
- Policy Enforcement: Strict guidelines for hand hygiene, lab coat usage, and biohazard waste disposal.
- Training & Education: Continuously training personnel on proper aseptic techniques and holding safety drills.
- Workflow Alteration: Grouping (cohorting) patients with the same infection together, or assigning dedicated nurses to only care for infected patients to prevent cross-ward contamination.
- Personal Protective Equipment (Protect the worker)
The least effective tier. PPE is the absolute final barrier between the pathogen and the healthcare worker. It relies 100% on perfect human compliance, perfect sizing/fit, and flawless technique (donning and doffing) to work.
III. Standard Precautions
Standard Precautions are the absolute minimum infection prevention practices that apply to ALL patient care, regardless of the suspected or confirmed infection status of the patient, in any setting where health care is delivered.
These practices are designed symmetrically: to protect the Healthcare Personnel (HCP) from the patient's flora, and to prevent the HCP from acting as a vector spreading infections among other patients.
- Hand hygiene.
- Use of Personal Protective Equipment (PPE).
- Respiratory hygiene / cough etiquette.
- Sharps safety.
- Safe injection practices.
IV. Focus: Hand Hygiene
Hand hygiene is universally recognized by the WHO and CDC as the single most important measure to prevent the spread of infections among patients and healthcare providers.
Methods of Hand Hygiene
- 1. Alcohol-Based Hand Rub (ABHR): The preferred, primary method for routine examinations and procedures when hands are NOT visibly soiled.
- Mechanism: Alcohol rapidly denatures microbial proteins, killing pathogens instantly.
- Advantages: It is much faster, more effective against most typical pathogens than soap, and contains emollients making it better tolerated by the skin during repetitive use.
- 2. Soap and Water (Hand Washing): Must be used when hands are visibly soiled (e.g., stained with dirt, blood, feces, or body fluids).
- Mechanism: Soap does not necessarily "kill" bacteria; instead, it is a surfactant. Combined with the mechanical friction of rubbing your hands, it physically lifts the microbes off the skin and washes them down the drain.
- 3. Surgical Hand Scrub: A prolonged, highly specific, rigorous surgical scrub (often using Chlorhexidine) that must be performed to eliminate transient flora and reduce resident flora before donning sterile surgeon's gloves for the OR.
The WHO "5 Moments for Hand Hygiene"
When should you clean your hands? Memorize these 5 critical moments:
- Before touching a patient (Protects the patient from the nurse's germs).
- Before a clean/aseptic procedure (Protects the patient from harmful germs entering their body, like inserting an IV).
- After body fluid exposure risk (Protects the nurse and the healthcare environment).
- After touching a patient (Protects the nurse from the patient's flora).
- After touching patient surroundings (Even if you only touched the bedrail or monitor, you must clean your hands before leaving).
The Soap and Water Exception: C. diff
Case: You have just finished examining a patient who is suffering from severe, watery diarrhea caused by Clostridioides difficile (C. diff). You wore gloves, and upon removing them, your hands are NOT visibly soiled. Should you use the Alcohol-Based Hand Rub (ABHR) or wash with soap and water?
Answer: You MUST wash with Soap and Water.
Why? Because C. diff (as well as Norovirus and Bacillus anthracis) forms highly resilient, hard-shelled spores. These spores are practically armor-plated and are highly resistant to the alcohol in ABHR. The alcohol will simply wet the spores without killing them. The only way to remove them is via the mechanical friction and rinsing action of soap and water, which physically washes the spores down the drain. This is a massive, highly-tested exception to standard hand hygiene rules!
V. Standard Precautions: Personal Protective Equipment (PPE)
Personal Protective Equipment (PPE) refers to wearable equipment specifically designed to protect HCP from exposure to or contact with infectious agents, as well as biological, chemical, radiological, or physical hazards.
Appropriate Use of PPE
- Gloves: Use in situations involving possible contact with blood, body fluids, mucous membranes, non-intact skin (e.g., rashes, open wounds), or heavily contaminated equipment.
- Protective Clothing (Gowns, Lab coats, Aprons): Use to protect intact skin and personal clothing during procedures or activities where splashing or contact with blood/body fluids is anticipated (e.g., changing a heavily soiled wound dressing, assisting in trauma).
- Mouth, Nose, and Eye Protection (Masks, Goggles, Face Shields): Use during procedures that are likely to generate splashes, sprays, or aerosols of blood or other body fluids (e.g., surgical procedures, dental work, suctioning an airway, or intubation).
Why target Mucous Membranes & OPIM?
Why are we so incredibly protective of mucous membranes (the wet pink tissues of the eyes, inside the nose, and the mouth)?
Unlike intact skin on your arm—which is covered by a thick, dry layer of dead, keratinized cells acting as a literal brick wall against bacteria—mucous membranes are living, wet, highly vascular tissues. They readily absorb fluids. If a droplet of infected blood splashes into your eye, the pathogen can be absorbed directly into your bloodstream within seconds.
Furthermore, infection guidelines constantly mention OPIM (Other Potentially Infectious Materials). While everyone knows blood is dangerous, OPIM encompasses a wide range of dangerous fluids requiring full PPE, including: Semen, vaginal secretions, cerebrospinal fluid (CSF), synovial fluid (joints), pleural fluid (lungs), pericardial fluid (heart), peritoneal fluid (abdomen), and amniotic fluid.
VI. Key Recommendations for PPE Usage
Merely providing PPE is insufficient; HCP must be rigorously trained on how to select, put on (don), remove (doff), and dispose of PPE safely without self-contaminating.
Rules for Gloves & Preventing Cross-Contamination
- Never reuse or wash gloves: Gloves are strictly single-use. Washing them with soap or alcohol degrades the latex/nitrile structural integrity instantly, creating microscopic holes you cannot see, rendering them useless.
- One patient per pair: Never wear the same pair of gloves for the care of more than one patient.
- Limit surface touching: Training must stress the dangers of cross-contamination. While wearing contaminated gloves, you must NOT touch clean environmental surfaces (e.g., do not type on the computer keyboard, adjust your glasses, answer your cell phone, or grab a door handle with bloody gloves!).
- Hand hygiene is the final step: You must perform hand hygiene immediately after removing gloves. Why? Because gloves develop micro-tears during use, and the warm, moist environment inside the glove acts as a bacterial incubator for flora on your hands.
The Strict Order of PPE Donning & Doffing
Improper PPE removal is the #1 way healthcare workers accidentally infect themselves (e.g., pulling a contaminated gown over your head and rubbing the infectious material into your eyes). Memorize these standard sequences:
DONNING (Putting On): Work from the bottom up, hands are always last.
- Gown: Fully cover torso from neck to knees.
- Mask or Respirator: Secure ties or elastic bands at middle of head and neck.
- Goggles or Face Shield: Place over face and eyes.
- Gloves: Extend to cover the wrist of the isolation gown.
DOFFING (Taking Off): The dirtiest items are removed first.
- Gloves: The most contaminated. Peel off carefully without snapping.
- Goggles or Face Shield: Handle only by the clean headband or earpieces.
- Gown: Unfasten ties, pull away from neck and shoulders, rolling it inside-out into a bundle.
- Mask or Respirator: Grasp bottom ties/elastics, then top, and remove without touching the contaminated front.
- Hand Hygiene: Wash hands immediately!
VII. Standard Precautions: Respiratory Hygiene / Cough Etiquette
These infection prevention measures are designed to limit the transmission of respiratory pathogens spread by droplet or airborne routes. They apply to patients, visitors, and HCP alike.
- Signage and Triage: Post visible signs at entrances instructing patients with respiratory symptoms (cough, runny nose) to alert staff immediately.
- Source Control: This is highly critical. Offer a surgical mask to coughing patients as soon as they enter the facility. Putting the mask directly on the patient traps the pathogen at the source before it can aerosolize into the waiting room.
- Spatial Separation: Provide space and encourage symptomatic persons to sit far away from others. The standard rule for basic droplet precautions is at least 3 to 6 feet (1 to 2 meters) of separation.
- Resources: Supply tissues, no-touch foot-pedal trash cans, and highly visible hand sanitizer stations in all waiting areas.
VIII. Standard Precautions: Sharps Safety
Needlestick injuries are a massive occupational hazard. Engineering and work-practice controls are the primary methods used to reduce exposures to bloodborne pathogens.
- Engineering Controls: Structurally isolating the hazard. Examples include heavy-duty, puncture-resistant red "Sharps Containers" mounted on walls, and syringes with built-in retractable safety shields.
- Work-Practice Controls: Changing human behavior. The absolute golden rule is: NEVER recap used needles using both hands.
- Physiology Expansion: Two-handed recapping is the leading cause of needle-stick injuries globally. If you hold the cap in one hand and the needle in the other, and you miss the tiny cap hole by a millimeter, you drive a hollow-bore needle filled with patient blood deeply into your own finger tissue.
- The Solution: If recapping is absolutely necessary (e.g., drawing up medication away from the bedside), use the One-Handed Scoop Technique—leave the cap on the table, scoop it up with the needle, and press it against a hard surface to secure it.
The Needle-Stick Injury Probability
Case: While cleaning up a chaotic procedure tray, a nurse accidentally pricks their finger deeply with a needle that was just used to draw blood from a patient. The patient's chart reveals a history of active Hepatitis B (HBV), Hepatitis C (HCV), and HIV. Which of these three bloodborne viruses is the nurse statistically most likely to contract from this single needle-stick injury?
Answer: Hepatitis B Virus (HBV).
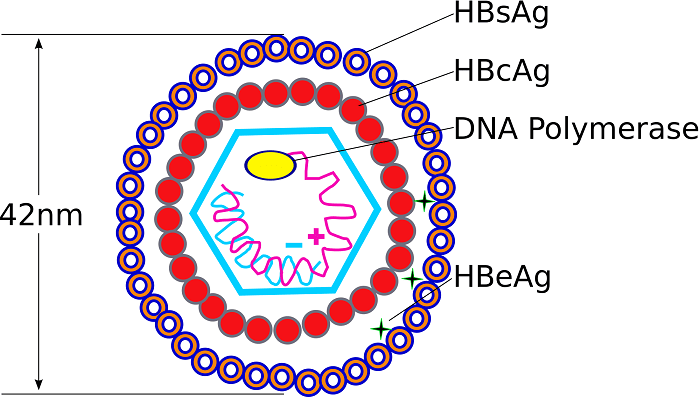
Rationale: The transmission risk is directly correlated to the viral load in the patient's blood and the hardiness of the virus. For a contaminated needle-stick, the risk of contracting HBV is terrifyingly high—up to 30% if the patient is highly infectious (HBeAg positive) and the HCP is unvaccinated. By contrast, the transmission risk for Hepatitis C is about 1.8%, and for HIV, the risk is surprisingly low at only about 0.3%. This high infectivity is exactly why the Hepatitis B vaccine series is strictly mandated for all clinical healthcare workers worldwide!
IX. Standard Precautions: Safe Injection Practices
Safe injection practices prevent the transmission of infectious diseases (specifically Hepatitis B, C, and HIV) between patients, or between a patient and HCP during parenteral (IV, IM, SubQ) medication administration.
Unsafe Practices Leading to Massive Patient Harm:
- Syringe Sharing: Administering medication to multiple patients using the same syringe. (Note: Even if you change the needle, the syringe is still contaminated! When you push fluid out, the release creates a microscopic vacuum effect that pulls invisible droplets of patient blood backward into the syringe barrel).
- Environmental Contamination: Preparing sterile IV medications in a "dirty" utility room or on a counter next to used blood tubes.
The Multi-Dose Vial Trap
One of the most insidious ways infections spread in clinics involves multi-dose vials (e.g., a large vial of Insulin or Lidocaine used for multiple patients).
Historically, massive outbreaks of Hepatitis C have been traced back to nurses making a fatal error: They use a brand new, sterile needle to draw medication for Patient A, but they attach it to a used syringe. By plunging the needle into the vial, the used syringe permanently contaminates the entire multi-dose vial with Patient A's Hepatitis C virus.
Later that day, a second nurse uses a completely new, sterile needle AND a sterile syringe to draw medication from that same vial for Patient B. Because the vial fluid itself is now infected, Patient B contracts the virus.
The Golden Rule of Injections: One Needle, One Syringe, One Time. Every single time you puncture a medication vial, it must be with a brand-new needle attached to a brand-new syringe.
X. Transmission-Based Precautions
While Standard Precautions apply to everyone, Transmission-Based Precautions represent the rigorous second tier of infection control. They are NEVER used alone; they are always implemented in addition to Standard Precautions for patients known or suspected to be infected with highly transmissible or epidemiologically important pathogens.
There are three specific categories based on the route of transmission: Contact, Droplet, and Airborne.
Used for infections spread by direct skin-to-skin contact with the patient, or indirect contact with highly contaminated environmental surfaces (bedrails, call buttons, blood pressure cuffs).
- Examples: MRSA, VRE, C. diff, Scabies, Shigella, Salmonella, RSV.
- PPE Required: Gown and Gloves donned immediately upon room entry.
- Equipment: Use dedicated, disposable patient-care equipment (e.g., leave a dedicated stethoscope and blood pressure cuff permanently inside the room).
- Patient Transport: Limit transport. Ensure any infected areas (wounds) are heavily bandaged and contained before moving the patient.
Designed to stop pathogens spread through close respiratory contact (sneezing, coughing, talking).
- The Physics: Droplets are large, "heavy" respiratory particles (typically >5 microns). Because of their weight, gravity pulls them down quickly. They travel a maximum of 3 to 6 feet before falling to the ground or landing on surfaces.
- PPE Required: Standard Surgical Mask (and eye protection if splashing is likely).
- Room: Single room preferred, but patients can be cohorted with others having the same active infection. No special ventilation required.
- Mnemonic: "PIMP my Ride"
- Pertussis (Whooping Cough)
- Influenza (Flu)
- Meningitis (Neisseria meningitidis)
- Pneumonia (Mycoplasma)
The highest level of respiratory isolation, used for highly contagious pathogens that survive in the air over long distances and time.
- The Physics: Airborne particles (droplet nuclei) are incredibly small, "light" particles (<5 microns). Evaporation turns them into microscopic dust. Like smoke, they defy gravity, remain suspended in the air for hours, and easily travel out into hallways and through HVAC ventilation systems.
- PPE Required: Fit-tested N95 Respirator (or higher PAPR). A standard surgical mask is absolutely useless, as air flows around the loose edges and the paper cannot filter sub-micron particles.
- Room: MUST be an AIIR (Airborne Infection Isolation Room). AIIRs feature Negative Air Pressure—air flows into the room from the hallway, never out. Air is exhausted directly outside or pushed through massive HEPA filters. The door must remain strictly closed.
- Mnemonic: "MTV"
- Measles (Rubeola)
- Tuberculosis (Pulmonary TB)
- Varicella (Chickenpox / Disseminated Shingles)
The Triage Decision
Case: A patient arrives at the clinic complaining of a chronic bloody cough, severe weight loss, and drenching night sweats. You suspect active Pulmonary Tuberculosis. You have two available rooms: a standard exam room and a room equipped with a negative-pressure ventilation system. Where do you place the patient, and what is your first immediate action?
Answer: Place the patient immediately in the Negative-Pressure Room (AIIR) and perform immediate Source Control by putting a standard surgical mask directly onto the patient's face.
Rationale: The mask traps the large infectious droplets as they exit the patient's mouth before they can evaporate into dangerous airborne droplet nuclei. The negative pressure room ensures that any "smoke-like" infectious particles they do exhale cannot draft out under the door and infect the immunocompromised patients sitting in the general waiting area.
XI. Care Bundles
Care bundles are a highly structured set of evidence-based guidelines, checklists, or interventions designed to dramatically improve the quality and safety of patient care.
- Definition & Philosophy: They typically consist of a small, manageable group (3 to 5) of critical interventions.
- The "Bundle" Principle (All-or-Nothing): Physiology dictates that these interventions work synergistically. When implemented together as a cohesive package, they have been shown to produce significantly better survival outcomes than when implemented individually or randomly. If a bundle has 5 required steps and the nurse only completes 4, the bundle is considered "failed," and the patient does not get the full protective benefit.
Examples of Critical Care Bundles
- Sepsis Bundles (The Hour-1 Bundle): Sepsis is a deadly blood infection causing organ failure. The bundle mandates that within the first hour, the team must: 1. Measure lactate levels. 2. Obtain blood cultures (before antibiotics). 3. Administer broad-spectrum antibiotics. 4. Begin rapid IV fluid resuscitation. 5. Apply vasopressors if blood pressure drops.
- Surgical Site Infection (SSI) Prevention Bundles: To prevent cutting open a patient and introducing bacteria, the bundle mandates: 1. Hair clipping instead of razor shaving (shaving creates microscopic skin cuts where bacteria breed). 2. Administering prophylactic IV antibiotics exactly 60 minutes prior to incision. 3. Maintaining strict blood glucose control during surgery.
- Ventilator-Associated Pneumonia (VAP) Bundles: Patients on life-support breathing machines easily get pneumonia. The bundle mandates: 1. Elevating the head of the bed 30-45 degrees (prevents acid reflux/aspiration into the lungs). 2. Daily "sedation vacations" (waking the patient up to see if they can breathe on their own). 3. Strict oral care scrubbing with chlorhexidine to kill mouth bacteria.
The Hierarchy of Controls & Checklists
Question: Why is a "checklist" considered an Administrative Control in the hierarchy of controls, and why is it absolutely essential for the success of Care Bundles?
Answer: A checklist is an Administrative Control because it fundamentally alters the "way people work" by enforcing a specific, written protocol and standardizing human behavior. It is essential for Care Bundles because bundles rely entirely on consistency and completeness. In a high-stress, fast-paced ICU environment, human memory is flawed. The checklist acts as an administrative safety net, ensuring that no single evidence-based step is forgotten, thereby maximizing patient survival.
List of References
- Centers for Disease Control and Prevention (CDC). (2020). Guidelines for Environmental Infection Control in Health-Care Facilities. Atlanta, GA: U.S. Department of Health and Human Services.
- World Health Organization (WHO). (2009). WHO Guidelines on Hand Hygiene in Health Care: First Global Patient Safety Challenge Clean Care is Safer Care. Geneva: World Health Organization.
- National Institute for Occupational Safety and Health (NIOSH). (2015). Hierarchy of Controls. U.S. Centers for Disease Control and Prevention.
- Siegel, J. D., Rhinehart, E., Jackson, M., & Chiarello, L. (2007). 2007 Guideline for Isolation Precautions: Preventing Transmission of Infectious Agents in Health Care Settings. American Journal of Infection Control.
- Institute for Healthcare Improvement (IHI). (2012). What is a Bundle? Cambridge, Massachusetts: IHI.
- Bennett, J. E., Dolin, R., & Blaser, M. J. (2019). Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases (9th ed.). Elsevier.
Quick Quiz
Infection Mitigation Quiz
Microbiology - mobile-friendly and focused practice.
Privacy: Your details are used only for quiz tracking and certificates.
Infection Mitigation Quiz
Microbiology
Preparing questions...
Choose your answer and keep your streak alive.
Great effort.
Here is your quick performance summary.
Infection Mitigation Measures Read More »