
Hepatobiliary System: Liver, Gallbladder
The Hepatobiliary System
The hepatobiliary system is a vital part of the digestive apparatus, comprising the liver, gallbladder, and the bile ducts. Its primary functions revolve around the production, transport, concentration, and regulated secretion of bile, which is crucial for lipid digestion and the excretion of waste products.
Main Functions:
- Bile Production: The liver continuously produces bile.
- Bile Transport: Bile ducts transport bile from the liver to the duodenum or to the gallbladder for storage.
- Bile Concentration: The gallbladder stores and concentrates bile.
- Bile Secretion: Bile is released into the duodenum to aid in digestion.
Bile Composition and Function:
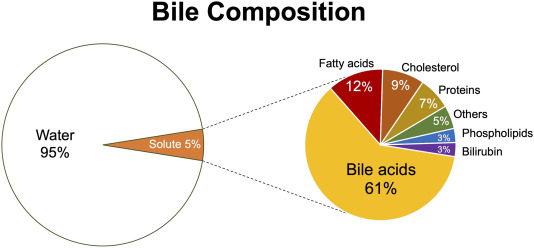
Bile is a complex, alkaline fluid (pH 7.6-8.6) typically yellowish-green in color, produced by hepatocytes. It consists of:
Bile Salts
Derived from cholesterol, these are the most functionally important component. They emulsify dietary fats (lipids and fatty acids) in the small intestine, breaking large fat globules into smaller ones. This increases the surface area for lipase enzymes to act upon, facilitating fat digestion and absorption (especially of fat-soluble vitamins A, D, E, K).
Bilirubin
A yellowish-green pigment, it is the primary waste product of heme metabolism (from the breakdown of old red blood cells). The liver conjugates bilirubin (makes it water-soluble) and excretes it into bile. Its presence gives feces their characteristic brown color (after further modification by gut bacteria).
Other Components
- Cholesterol: A significant component of bile, primarily excreted this way.
- Phospholipids (e.g., lecithin): Aid in emulsification.
- Water: The largest component.
- Electrolytes: Bicarbonate, sodium, potassium, chloride.
- Trace metals, heavy metals, drugs: Excreted via bile.
The Liver
The liver is the largest gland in the body and also the largest internal organ, weighing approximately 1.3-1.5 kg (around 2% of adult body weight). It is a metabolically diverse and highly vascularized organ with an astounding array of functions, far beyond just bile production.
Location and Structure
- Location: Occupies most of the upper right quadrant of the abdomen, extending into the epigastric region and even into the upper left quadrant. It lies immediately inferior to the diaphragm, largely protected by the lower ribs.
- Surfaces:
- Diaphragmatic (Anterior/Superior) Surface: This large, convex surface is molded by the diaphragm.
- Visceral (Postero-inferior) Surface: This irregular, concave surface is molded by the various abdominal organs it contacts (e.g., stomach, duodenum, colon, right kidney, adrenal gland).
Basic Functions (Beyond bile production):
While bile production is a key excretory function, the liver's metabolic roles are paramount:
- Metabolic Activities: Central to the metabolism of:
- Carbohydrate Metabolism: Maintenance of blood glucose homeostasis Glycogenesis (glucose to glycogen), glycogenolysis (glycogen to glucose), gluconeogenesis (non-carbohydrate sources to glucose), regulation of blood glucose.
- Lipid Metabolism (Fats): Synthesis and degradation of fatty acids, cholesterol, triglycerides, and lipoproteins. Synthesis of cholesterol, lipoproteins, triglycerides; fatty acid oxidation; ketone body formation.
- Protein Metabolism: Synthesis of most plasma proteins (e.g., albumin, clotting factors), deamination of amino acids, urea formation (detoxification of ammonia). Deamination of amino acids, leading to the formation of urea (detoxification of ammonia).
- Detoxification and Filtration:
- Detoxification: Metabolizes and detoxifies various endogenous substances (e.g., hormones) and exogenous substances (e.g., drugs, toxins, alcohol).
- Filtration: Its extensive vascular network (hepatic sinusoids) and specialized macrophages (Kupffer cells) act as a reticuloendothelial system, filtering blood to remove bacteria, foreign particles, old red blood cells, and cellular debris that have gained entry through the gastrointestinal tract via the portal circulation.
- Storage: Stores glycogen, vitamins (A, D, B12), iron (ferritin), and copper.
- Synthesis of Blood Components:
- Plasma Proteins: Albumin (maintains oncotic pressure), globulins, fibrinogen, prothrombin, and other clotting factors.
- Heparin: While often listed, heparin is primarily produced by mast cells and basophils. The liver's role is more in the synthesis of other anticoagulants and factors involved in coagulation.
- Production and Secretion of Bile: Essential for fat digestion and absorption, and for the excretion of waste products like bilirubin. Bilirubin is a breakdown product of heme (from worn-out red blood cells' hemoglobin), which the liver processes and excretes into bile.
- Immunological Role: Contains Kupffer cells (macrophages) which phagocytose foreign material and play a role in immune surveillance.
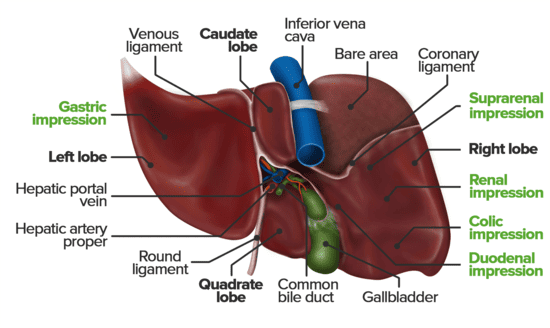
Lobes of the Liver
Traditionally, the liver is divided into four anatomical lobes based on external landmarks:
- Right Lobe: The largest lobe, occupying the right side of the liver. It is demarcated from the left lobe by the falciform ligament on the anterior/superior surface. On the visceral surface, it includes the gallbladder fossa and groove for the inferior vena cava.
- Left Lobe: Smaller than the right lobe, located to the left of the falciform ligament.
- Caudate Lobe: A small, quadrilateral lobe located on the visceral surface, superior to the porta hepatis, between the fissure for the ligamentum venosum (left) and the groove for the inferior vena cava (right).
- Quadrate Lobe: A small, quadrilateral lobe located on the visceral surface, inferior to the porta hepatis, between the fissure for the ligamentum teres (left) and the gallbladder fossa (right).
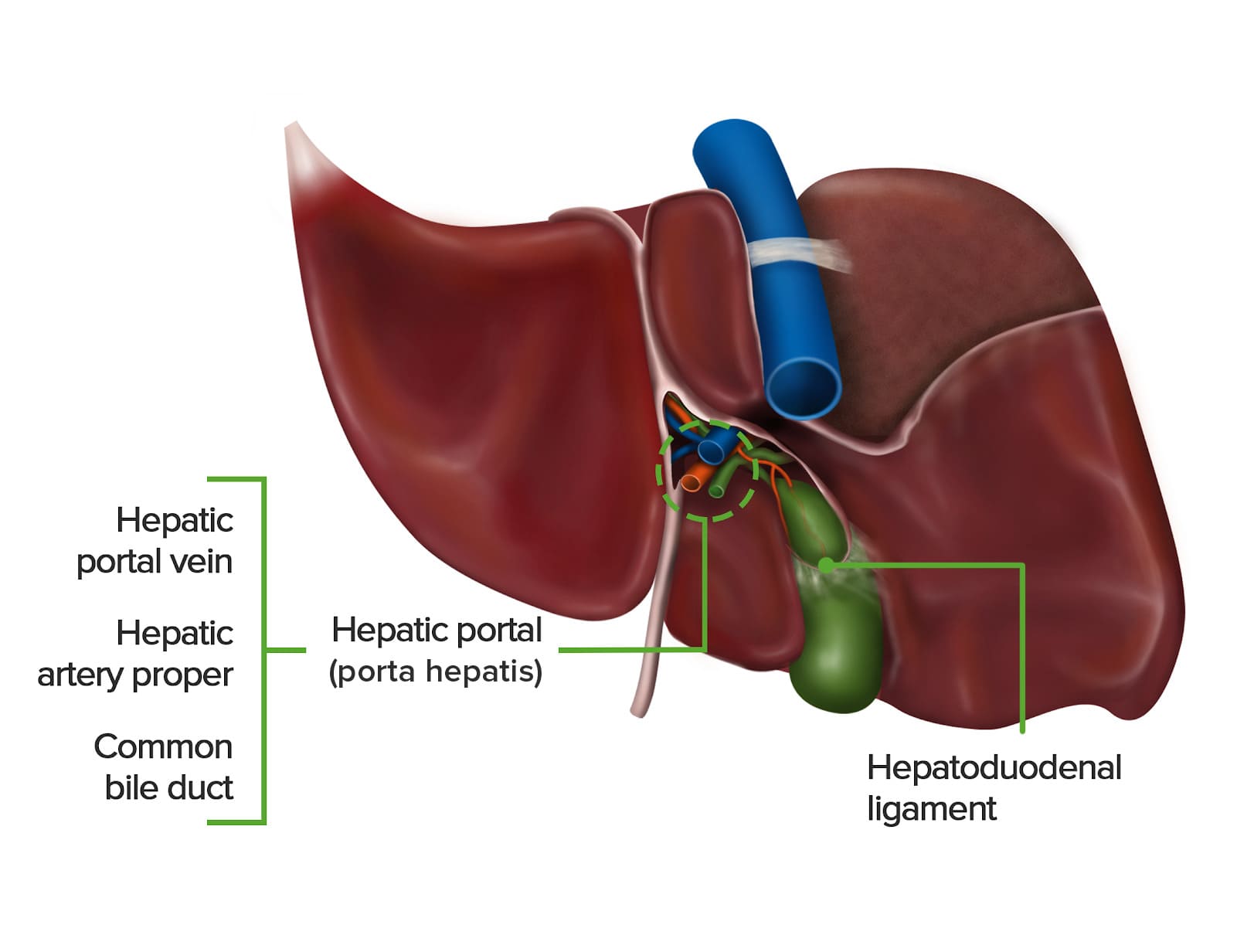
Porta Hepatis (Liver Hilum)
- Location: Found on the visceral (postero-inferior) surface of the liver. It is a deep transverse fissure situated between the caudate lobe superiorly and the quadrate lobe inferiorly.
- Attachment: The lesser omentum (specifically, the hepatoduodenal ligament part) attaches to its margins.
- Contents (Portal Triad and associated structures): The porta hepatis is the entry and exit point for all structures forming the "portal triad" and other associated elements:
- Proper Hepatic Artery: Divides into right and left hepatic arteries, which supply oxygenated blood to the liver parenchyma.
- Hepatic Portal Vein: Divides into right and left portal veins, carrying nutrient-rich, deoxygenated blood from the gastrointestinal tract, spleen, and pancreas to the liver for processing.
- Common Hepatic Duct: Formed by the union of the right and left hepatic ducts, which drain bile from the liver.
- Lymphatic Vessels: Drain lymph from the liver.
- Nerve Fibers: Sympathetic and parasympathetic nerve fibers (from the hepatic plexus), which regulate liver function and blood flow.
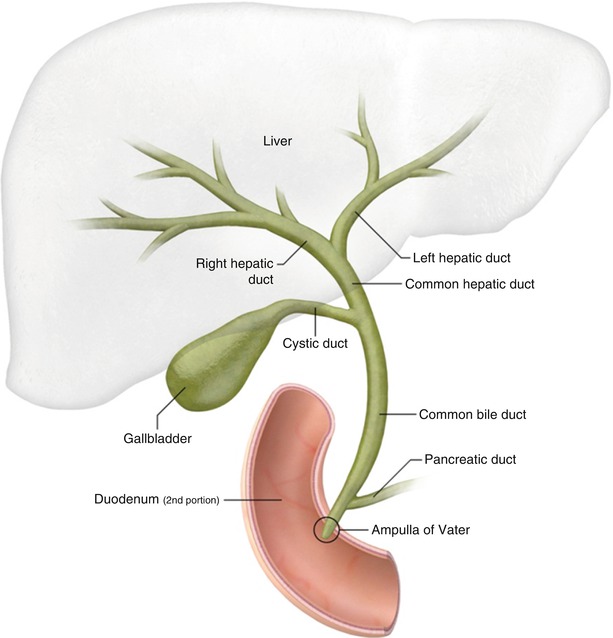
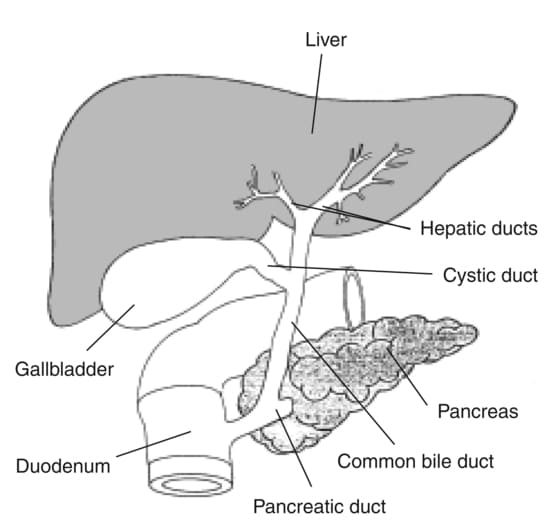
The Biliary Ducts
The biliary tree is a system of ducts that transports bile from the liver to the duodenum.
Intrahepatic Bile Ducts:
Small bile canaliculi collect bile from hepatocytes, forming progressively larger ductules within the liver. These eventually merge to form the right and left hepatic ducts.
Extrahepatic Bile Ducts:
- Right Hepatic Duct: Drains bile from the right anatomical/functional lobes.
- Left Hepatic Duct: Drains bile from the left, caudate, and quadrate lobes.
- Common Hepatic Duct (CHD): Formed by the union of the right and left hepatic ducts, typically just outside the porta hepatis.
- Cystic Duct: Originates from the gallbladder and connects to the common hepatic duct. It has spiral folds of mucosa (spiral valve of Heister) that help keep the duct open and regulate bile flow in and out of the gallbladder.
- Common Bile Duct (CBD): Formed by the union of the common hepatic duct and the cystic duct. It descends posterior to the first part of the duodenum.
- Pancreatic Duct (of Wirsung): The main duct draining the pancreas, carrying pancreatic enzymes.
- Hepatopancreatic Ampulla (Ampulla of Vater): The common bile duct usually joins the main pancreatic duct to form a short, dilated common channel called the ampulla of Vater.
- Major Duodenal Papilla: The ampulla of Vater opens into the second part of the duodenum via this small, raised nipple-like structure.
- Sphincter of Oddi (Hepatopancreatic Sphincter): A complex smooth muscle sphincter surrounding the ampulla of Vater (and sometimes individually surrounding the distal common bile duct and pancreatic duct). It regulates the flow of bile and pancreatic juice into the duodenum and prevents reflux of duodenal contents.
Bile Ducts (Referred to as the Biliary Tree)
Bile, produced continuously by hepatocytes, is secreted into a complex system of ducts that transport it to the duodenum or gallbladder.
- Bile Flow Rate: The liver produces bile at an average rate of 400-800 ml per day (not 40 ml/hour continuously, which would be an extremely high volume). It is then stored and concentrated in the gallbladder.
Intrahepatic Ducts:
- Bile Canaliculi: Small channels between hepatocytes.
- Bile Ductules (Canals of Hering): Small ducts that collect bile from canaliculi.
- Interlobular Bile Ducts: Found in the portal triads, these ducts receive bile from the ductules. They gradually merge to form progressively larger ducts within the liver.
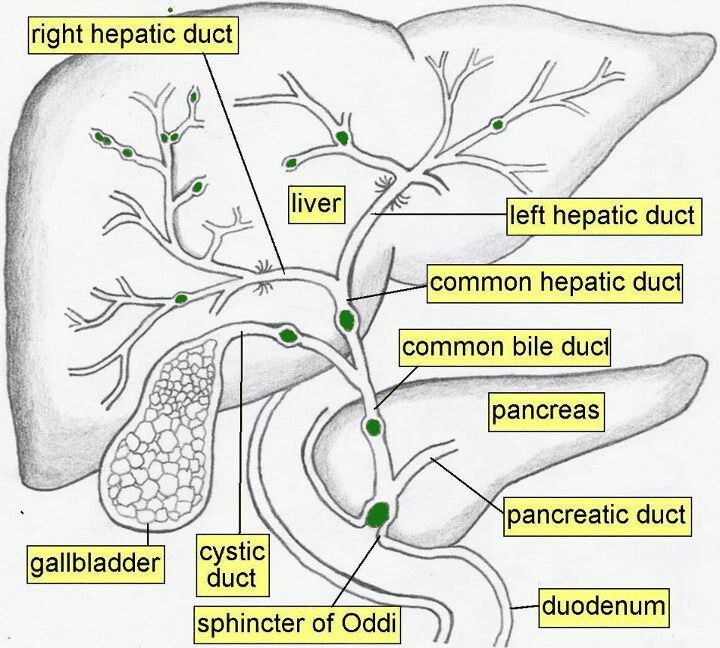
Extrahepatic Ducts:
- Right and Left Hepatic Ducts: The progressively larger intrahepatic ducts eventually emerge from the porta hepatis as the right and left hepatic ducts, draining bile from their respective liver territories.
- Common Hepatic Duct (CHD): Formed by the union of the right and left hepatic ducts.
- Length: Approximately 3 cm long (not 4 cm, although there can be slight variations).
- Course: Descends in the free margin of the lesser omentum.
- Cystic Duct: Connects the gallbladder to the common hepatic duct.
- Common Bile Duct (CBD) / Bile Duct: Formed by the union of the common hepatic duct and the cystic duct.
- Length: Approximately 7-10 cm long (8 cm is a good average).
- Course: Divided into three main parts:
- Supraduodenal (First) Part: Lies in the free margin of the lesser omentum, anterior to the portal vein and to the right of the hepatic artery proper. This is generally the most accessible portion surgically.
- Retroduodenal (Second) Part: Lies posterior to the first part of the duodenum.
- Infraduodenal (Third) Part: Descends in a groove or tunnel on the posterior surface of the head of the pancreas, usually lying anterior to the right renal vein (not behind it). Here, it typically joins the main pancreatic duct (of Wirsung).
- Termination: The common bile duct and main pancreatic duct usually unite to form the hepatopancreatic ampulla (Ampulla of Vater), which opens into the second part of the duodenum at the major duodenal papilla. The flow of bile and pancreatic juice is regulated by the sphincter of Oddi.
Histology of the Liver
The liver's unique histological organization is fundamental to its diverse functions.
Connective Tissue Framework:
- The entire liver is surrounded by a dense fibrous capsule known as Glisson's capsule.
- Bare Area: The only region of the liver not covered by visceral peritoneum is the bare area on the posterosuperior diaphragmatic surface. Here, Glisson's capsule is in direct contact with the diaphragm. This area is bounded by the superior and inferior layers of the coronary ligament.
- At the porta hepatis (hilum), Glisson's capsule invaginates into the liver substance, sending connective tissue septa (portal tracts) throughout the organ. These septa provide structural support and create channels for the passage of blood vessels, bile ducts, lymphatic vessels, and nerves.
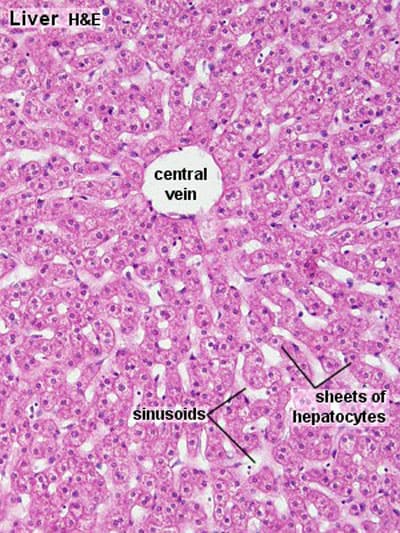
Liver Lobules – Functional Units
The liver is histologically organized into functional units, primarily described by three models: the classic hepatic lobule, the portal lobule, and the liver acinus. The classic hepatic lobule is the most commonly described for general anatomical understanding.
1. Classic Hepatic Lobule:
- Shape: Typically depicted as an irregular hexagonal prism.
- Boundaries: Defined by sparse collagenous connective tissue containing portal triads (or portal canals) at its corners.
- Central Vein (Terminal Hepatic Venule): Each lobule has a central vein (also called the terminal hepatic venule) located at its center. This vein is a tributary of the hepatic veins, which ultimately drain into the inferior vena cava.
- Hepatocytes (Liver Cells): Radiate outwards from the central vein in anastomosing plates or cords, like spokes of a wheel. These cords are typically one or two cells thick.
- Hepatic Sinusoids: Located between the plates of hepatocytes are dilated, discontinuous capillaries called hepatic sinusoids. These receive blood from both the hepatic artery and the portal vein at the periphery of the lobule.
- Blood Flow: Blood flows from the portal triads at the periphery of the lobule, through the sinusoids, and converges towards the central vein.
- Cellular Components of Sinusoids:
- Endothelial Cells: Form the lining, but with large fenestrations and gaps, allowing plasma to directly contact hepatocytes.
- Kupffer Cells: Specialized macrophages derived from monocytes, found within the sinusoids. They phagocytose foreign material, bacteria, and old red blood cells, playing a crucial role in the liver's immune defense and blood filtration.
- Hepatic Stellate Cells (Ito cells): Located in the Space of Disse (the perisinusoidal space between endothelial cells and hepatocytes). These cells store vitamin A and, in pathological conditions, can transform into myofibroblasts, producing collagen and contributing to liver fibrosis.
- Space of Disse: The perisinusoidal space between the sinusoidal endothelium and the hepatocytes. It allows direct exchange of materials between plasma and hepatocytes.
2. Portal Triad (Portal Canal):
- Located at the corners of the classic hepatic lobule, embedded within the connective tissue septum.
- Consists of three main structures:
- Branch of the Hepatic Artery: Supplies oxygenated blood (and bile duct epithelium) to the lobule.
- Branch of the Hepatic Portal Vein: Supplies nutrient-rich, deoxygenated blood to the lobule.
- Bile Ductule (Interlobular Bile Duct): A small duct that collects bile produced by hepatocytes.
- Also contains lymphatic vessels and nerves.
Functions of Hepatocytes:
Hepatocytes are remarkably versatile cells, performing a vast array of metabolic, synthetic, and detoxification functions:
- Synthesis of Bile: Produce bile salts, cholesterol, phospholipids, and other components of bile.
- Metabolic Processing:
- Carbohydrate Metabolism: Glycogenesis, glycogenolysis, gluconeogenesis, glucose regulation.
- Lipid Metabolism: Synthesis of lipoproteins, cholesterol, triglycerides; fatty acid oxidation.
- Protein Metabolism: Synthesis of plasma proteins (e.g., albumin, clotting factors, acute phase proteins), deamination of amino acids, urea synthesis (detoxification of ammonia).
- Detoxification and Biotransformation: Metabolize and inactivate drugs, toxins, hormones (e.g., estrogen, glucocorticoids) through oxidation, reduction, hydrolysis, and conjugation reactions.
- Storage: Store glycogen, fat, iron (as ferritin), and fat-soluble vitamins (A, D, B12).
- Immune Function: Work with Kupffer cells to present antigens and clear immune complexes.
Relations of the Liver
The liver has extensive relations with surrounding structures due to its large size and position.
Anterior Relations:
- Diaphragm
- Right and left costal margins (lower ribs)
- Right and left pleura and the lower margins of both lungs (during full inspiration)
- Xiphoid process
- Anterior abdominal wall
Posterior Relations (Visceral Surface):
- Diaphragm
- Right kidney (superior pole) and right suprarenal gland
- Hepatic flexure of the colon
- First part of the duodenum
- Gallbladder
- Inferior vena cava (often partially embedded in the liver)
- Esophagus (abdominal part)
- Fundus and body of the stomach (imprint on the left lobe)
- Lesser omentum
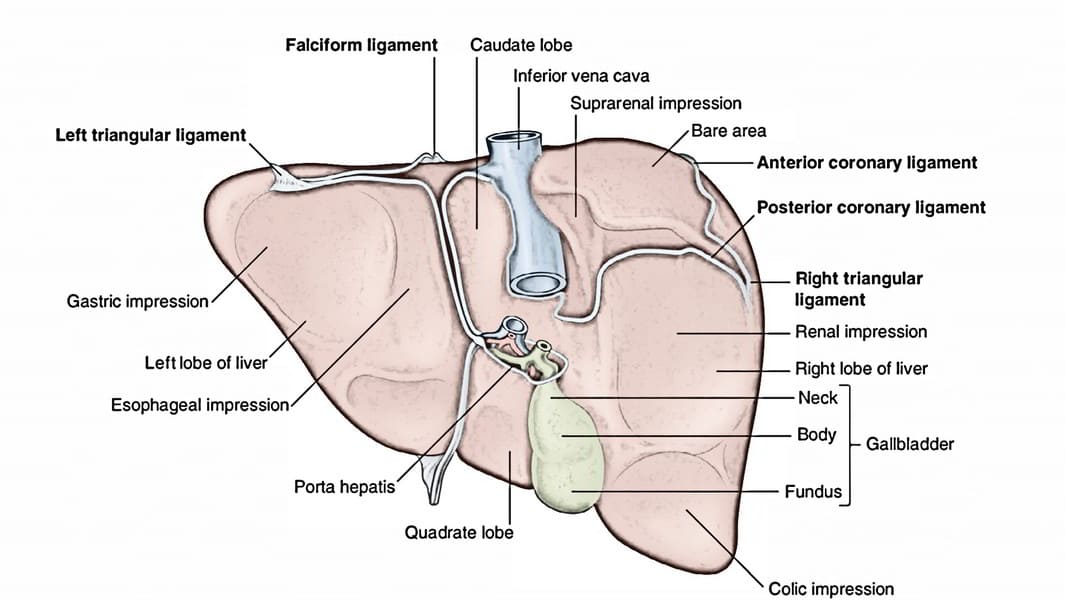
Ligaments of the Liver
The liver is held in place by several peritoneal folds and remnants of fetal structures, collectively referred to as ligaments.
Falciform Ligament
Description: A sickle-shaped, two-layered fold of peritoneum that attaches the liver to the anterior abdominal wall (from the umbilicus superiorly) and the undersurface of the diaphragm.
Location: Found on the supero-anterior surface of the liver, dividing the anatomical right and left lobes.
Contents: Its lower, free margin contains the ligamentum teres hepatis (round ligament of the liver).
Development: A remnant of the ventral mesentery.
Ligamentum Teres Hepatis (Round Ligament of the Liver)
Description: A fibrous cord in the free lower edge of the falciform ligament.
Development: The obliterated remnant of the umbilical vein, which carried oxygenated blood from the placenta to the fetal liver.
Coronary Ligament
Description: Peritoneal reflections from the diaphragmatic surface of the liver to the inferior surface of the diaphragm.
Layers: It has a superior layer and an inferior layer, which define the boundaries of the bare area of the liver.
Continuations: Laterally, the layers of the coronary ligament come together to form the triangular ligaments.
- Right Triangular Ligament: Formed by the union of the right superior and inferior layers of the coronary ligament.
- Left Triangular Ligament: Formed by the union of the left superior and inferior layers of the coronary ligament.
Bare Area of the Liver
Description: This is the area on the posterior surface of the liver that is not covered by visceral peritoneum.
Boundaries: It is situated between the superior and inferior layers of the coronary ligament.
Clinical Significance: It allows direct contact between the liver and the diaphragm, and through this area, the inferior vena cava passes. This region is a potential site for infection to spread between the abdominal cavity and the thoracic cavity.
Lesser Omentum (Hepatogastric and Hepatoduodenal Ligaments)
Description: A double-layered peritoneal fold connecting the lesser curvature of the stomach and the first part of the duodenum to the liver.
- Hepatogastric Ligament: Connects the lesser curvature of the stomach to the liver.
- Hepatoduodenal Ligament: The free right border of the lesser omentum. It is important as it contains the portal triad (proper hepatic artery, hepatic portal vein, and common bile duct), along with lymphatic vessels and nerves, as it enters the porta hepatis.
Ligamentum Venosum
Description: A fibrous remnant embedded within the fissure for the ligamentum venosum on the visceral surface of the liver.
Development: The obliterated remnant of the ductus venosus, a fetal shunt that allowed umbilical venous blood to bypass the hepatic sinusoids and directly enter the inferior vena cava, thus bypassing much of the liver's metabolic activity.
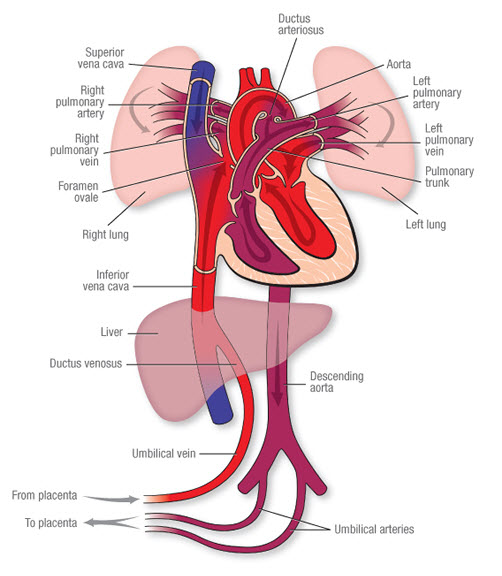
Fetal Liver Circulation
Fetal circulation is uniquely adapted to bypass the lungs and largely bypass the liver, as the placenta performs the functions of gas exchange and nutrient processing.
- Oxygenated Blood Delivery: In the fetus, highly oxygenated, nutrient-rich blood from the placenta is brought to the fetus via the umbilical vein.
- Liver Bypass (Ductus Venosus): Upon entering the fetal abdomen, the umbilical vein ascends to the liver.
- A smaller proportion of this blood supplies the fetal liver parenchyma.
- The greater proportion of the oxygenated blood bypasses the liver sinusoids by shunting directly into the inferior vena cava (IVC) via the ductus venosus. This ensures that the most oxygenated blood reaches the fetal heart (and subsequently the brain).
- Postnatal Changes:
- At birth, with the cessation of placental blood flow and the onset of pulmonary respiration, these fetal shunts are no longer needed.
- The umbilical vein gradually occludes and becomes the ligamentum teres hepatis (round ligament of the liver).
- The ductus venosus also closes functionally within hours of birth and anatomically within days/weeks, forming the ligamentum venosum. These are crucial anatomical landmarks in the adult liver.
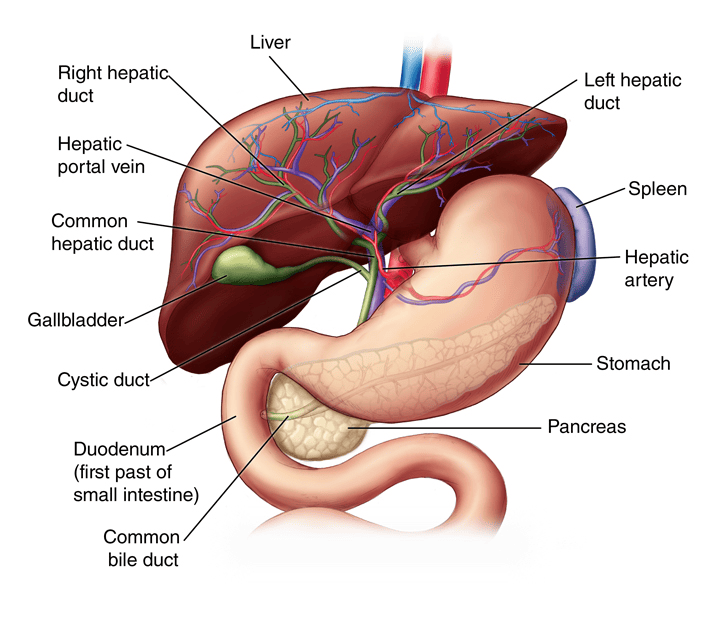
Blood Supply of the Liver
The liver has a unique dual blood supply, receiving both oxygenated and deoxygenated blood.
1. Hepatic Artery Proper (Oxygenated Blood):
- Origin: The common hepatic artery is a branch of the celiac trunk. It then gives off the gastroduodenal artery and continues as the hepatic artery proper.
- Course: The hepatic artery proper ascends in the hepatoduodenal ligament (part of the lesser omentum), anterior to the portal vein and to the left of the common bile duct.
- Branches: At the porta hepatis, it typically divides into the right hepatic artery and left hepatic artery, which then further branch to supply the respective functional lobes and segments of the liver. The right hepatic artery usually gives off the cystic artery to the gallbladder.
- Function: Supplies approximately 25-30% of the total blood flow to the liver, providing the liver parenchyma with oxygenated blood.
2. Hepatic Portal Vein (Deoxygenated, Nutrient-Rich Blood):
- Formation: Formed by the confluence of the superior mesenteric vein and the splenic vein posterior to the neck of the pancreas. It also receives blood from the inferior mesenteric vein (often via the splenic vein) and gastric veins.
- Course: Ascends in the hepatoduodenal ligament, posterior to the hepatic artery proper and common bile duct.
- Branches: At the porta hepatis, it typically divides into the right portal vein and left portal vein, which supply the respective functional lobes and segments.
- Function: Supplies approximately 70-75% of the total blood flow to the liver. This blood is rich in nutrients absorbed from the gastrointestinal tract and contains metabolic byproducts and toxins from the spleen and pancreas, all destined for processing by the liver.
Intrahepatic Circulation: Within the liver, branches of the hepatic artery and portal vein both terminate in the hepatic sinusoids. This mixed blood then flows through the sinusoids, comes into contact with hepatocytes, and drains into the central veins (terminal hepatic venules) of the hepatic lobules.
3. Hepatic Veins (Venous Drainage from the Liver):
- Formation: The central veins of all hepatic lobules coalesce into progressively larger sublobular veins, which eventually form the hepatic veins.
- Number and Course: There are typically three major hepatic veins: right, middle, and left. They emerge from the posterior surface of the liver and drain directly into the inferior vena cava (IVC), just below the diaphragm.
- Function: Carry deoxygenated, processed blood away from the liver back to the systemic circulation.
Lymphatic Drainage of the Liver
The liver is one of the most prolific lymph-producing organs in the body, generating about one-third to one-half of all lymph formed in the body.
- Superficial Lymphatics: Located beneath the capsule.
- Lymph from the superficial surfaces of the liver drains towards lymph nodes in the porta hepatis, eventually reaching the celiac lymph nodes.
- From the bare area of the liver, lymph vessels pierce the diaphragm and drain into lymph nodes in the posterior mediastinum (e.g., posterior mediastinal nodes, phrenic nodes).
- Deep Lymphatics: Located within the liver parenchyma.
- Follow the portal triads and hepatic veins.
- Most deep lymphatics ultimately drain towards lymph nodes in the porta hepatis. From there, lymph typically flows to the celiac lymph nodes, and then to the cisterna chyli and thoracic duct.
Nerve Supply of the Liver
The liver receives both sympathetic and parasympathetic innervation, primarily from the hepatic plexus.
- Hepatic Plexus: A network of nerves surrounding the hepatic artery and portal vein, derived from the celiac plexus.
- Sympathetic Innervation:
- Origin: Greater and lesser splanchnic nerves (T7-T9 segments of the spinal cord).
- Function: Primarily causes vasoconstriction of hepatic blood vessels, potentially reducing blood flow (though less significant than portal pressure changes). May also inhibit bile flow.
- Parasympathetic Innervation:
- Origin: Anterior and posterior vagal trunks (branches of the vagus nerve, CN X).
- Function: Primarily causes vasodilation (though also less significant than portal pressure changes). Stimulates bile formation (choleretic effect).
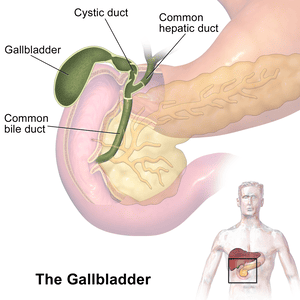
Gallbladder
The gallbladder is a small, hollow organ crucial for the storage and concentration of bile.
- Location: An intraperitoneal organ (except for the area where it directly contacts the liver) situated in a fossa on the visceral (inferior) surface of the right lobe of the liver.
- Shape: A pear-shaped organ nestled in a fossa on the visceral surface of the right lobe of the liver.
- Main Function: Stores and concentrates bile (by absorbing water and electrolytes) produced by the liver. Its capacity is typically 30-60 ml (50 ml is a good average). It removes water and electrolytes from bile, making it up to 5-10 times more concentrated.
- Parts:
- Fundus: The broadest, most distal part. It projects beyond the inferior border of the liver and often meets the anterior abdominal wall at the tip of the right ninth costal cartilage (at the junction of the lateral border of the rectus abdominis and the costal margin).
- Body: The main part of the gallbladder, lying in contact with the visceral surface of the liver, directed upwards, backwards, and to the left.
- Neck: The narrow, tapering end that is continuous with the cystic duct. It often has a pouch-like dilation called Hartmann's pouch, a common site for gallstone impaction.
- Regulation of Bile Release: When fatty food enters the duodenum, it stimulates the release of the hormone cholecystokinin (CCK). CCK causes the gallbladder to contract and the sphincter of Oddi to relax, allowing concentrated bile to flow into the duodenum.
- Relations:
- Anteriorly: Visceral surface of the liver, anterior abdominal wall (at the fundus).
- Posteriorly: Transverse colon, first part of the duodenum (and sometimes the second part).
- Blood Supply of the Gallbladder:
- Arterial Supply: Primarily by the cystic artery, which is usually a branch of the right hepatic artery.
- Venous Drainage: The cystic veins generally drain directly into the portal vein or into the liver sinusoids directly.
- Lymphatic Drainage: Similar to the liver, lymphatic vessels drain towards the lymph nodes in the porta hepatis and then to the celiac lymph nodes.
Cystic Duct
- Length: Approximately 3-4 cm long (not 8 cm).
- Course: Connects the neck of the gallbladder to the common hepatic duct to form the common bile duct. It descends in the free margin of the lesser omentum.
- Spiral Valve (Valves of Heister): Contains prominent spiral folds of mucosa that project into the lumen. These folds help keep the lumen patent, prevent sudden distension or collapse of the duct, and regulate bile flow in and out of the gallbladder. They are not true valves but rather mucosal folds.
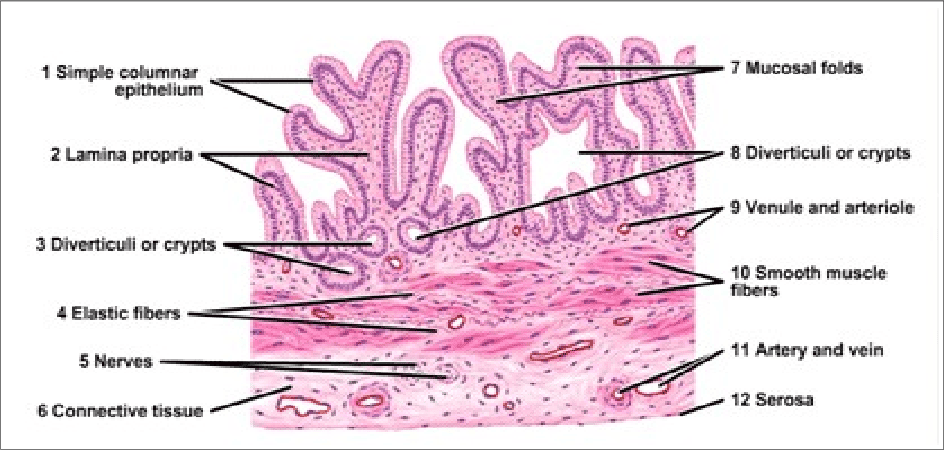
Histology of the Gallbladder
The gallbladder wall is adapted for its functions of storage and concentration.
- Mucosa:
- Epithelium: Lined by simple columnar epithelium with microvilli, specialized for absorbing water and electrolytes from bile.
- Folds: The mucosa is highly folded, especially when the gallbladder is empty, giving it a rugated appearance. These folds disappear when the gallbladder is distended. No goblet cells are typically present.
- Lamina Propria: A loose connective tissue layer beneath the epithelium, containing capillaries and lymphatic vessels involved in water absorption.
- Muscularis Externa (Muscular Layer): Composed of an oblique layer of smooth muscle fibers (rather than distinct inner circular and outer longitudinal layers found in most GI tract organs). Contraction of this muscle layer, stimulated by CCK, expels bile.
- Serosa/Adventitia:
- Serosa: The outer layer covering the free surface of the gallbladder, consisting of mesothelium and a thin layer of connective tissue (intraperitoneal surface).
- Adventitia: Where the gallbladder is attached directly to the liver (hepatic surface), it lacks a serosa and has an adventitia, which is a layer of dense irregular connective tissue.
- Absence of Submucosa and Muscularis Mucosae: The gallbladder typically lacks a distinct submucosa and muscularis mucosae, unlike other parts of the gastrointestinal tract. The lamina propria directly contacts the muscularis externa.
- Rokitansky-Aschoff Sinuses: Invaginations of the epithelial lining through the muscular layer, often extending into the subserosal connective tissue. These are common but can be pathological if excessively deep, potentially harboring bacteria and contributing to inflammation.
Embryology of the Hepatobiliary System
The development of the liver, gallbladder, and bile ducts is a complex process primarily originating from the embryonic foregut endoderm and interacting with surrounding mesoderm.
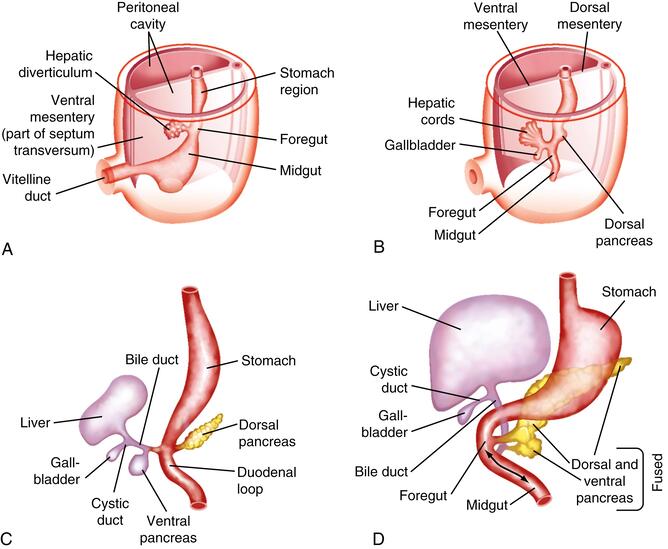
Early Stages (Week 3-4 IUL):
- Hepatic Diverticulum (Liver Bud):
- Development begins during the 3rd week of intrauterine life (IUL).
- It appears as a ventral outgrowth (diverticulum) of the endodermal epithelium at the distal end of the foregut (which will eventually become the duodenum).
- This outgrowth is often referred to as the hepatic diverticulum or liver bud.
- It consists of rapidly proliferating endodermal cells, which are the precursor cells for hepatocytes and bile duct epithelial cells (hepatoblasts).
- Penetration of the Septum Transversum:
- The proliferating hepatic diverticulum cells invade the septum transversum. The septum transversum is a block of splanchnic mesoderm that will eventually contribute to the diaphragm and ventral mesentery.
- This mesoderm is crucial, as it induces and interacts with the endodermal cells, guiding liver development.
Differentiation and Organogenesis (Week 4-12 IUL):
- Formation of the Bile Duct: As the liver bud continues to proliferate and invade the septum transversum, the connection between the hepatic diverticulum and the foregut (duodenum) gradually narrows. This narrowing stalk differentiates into the bile duct (common bile duct).
- Cystic Diverticulum and Gallbladder Formation:
- A small, secondary outgrowth (diverticulum) develops from the ventral aspect of the developing bile duct.
- This cystic diverticulum subsequently differentiates into the gallbladder and its connecting duct, the cystic duct.
- Formation of Hepatic Cords and Sinusoids:
- The proliferating endodermal cells within the developing liver form branching hepatic cords (precursors to hepatic plates/cords).
- These hepatic cords intermingle and anastomose with the vitelline and umbilical veins, which are already present within the septum transversum.
- The endothelial lining of these embryonic veins (vitelline and umbilical) gives rise to the hepatic sinusoids. This intimate relationship between hepatic cords and sinusoids is critical for the liver's circulatory function.
- Differentiation of Hepatoblasts:
- The original endodermal cells of the hepatic diverticulum (hepatoblasts) differentiate into:
- Hepatocytes (Parenchymal cells of the liver): The main functional cells of the liver.
- Cholangiocytes (Bile Duct Lining Cells): Form the lining of the entire intrahepatic and extrahepatic biliary tree. This process involves the formation of the ductal plate, a double-layered cylindrical structure of cholangiocytes around a mesodermal core. Remodeling of the ductal plate leads to the mature intrahepatic bile ducts.
- The original endodermal cells of the hepatic diverticulum (hepatoblasts) differentiate into:
- Mesodermal Contributions: While hepatocytes and cholangiocytes are endodermal, other important liver cells are of mesodermal origin:
- Kupffer cells: Derived from monocytes originating in the yolk sac and fetal liver mesoderm.
- Hepatic stellate cells (Ito cells): Derived from mesenchyme within the septum transversum.
- Connective tissue cells: Fibroblasts and other stromal cells that form the capsule and supporting framework.
- Hematopoietic cells: The fetal liver is a major site of hematopoiesis (blood cell formation) during embryonic and early fetal life.
Fetal Liver Functions and Growth:
- Hematopoiesis:
- Hematopoiesis begins in the liver around the 6th week IUL and becomes a prominent function, peaking around the 10th week IUL.
- At this stage, the liver is disproportionately large, making up approximately 10% of the total fetal body weight.
- The liver remains the primary site of blood cell formation until about 6-7 months of gestation, after which the bone marrow takes over.
- By birth, the hematopoietic function of the liver significantly reduces, and its relative size decreases to about 5% of total body weight.
- Bile Production:
- Bile formation by fetal hepatocytes begins around the 12th week IUL.
- Once secreted, bile enters the gastrointestinal tract and contributes to the formation of meconium, the dark green, sterile first stool of a newborn, which is primarily composed of desquamated epithelial cells, intestinal secretions, and bile.
| Component | Embryonic Origin | Differentiated Cells |
|---|---|---|
| Endoderm | Hepatic Diverticulum / Liver Bud | Hepatocytes, Biliary Canaliculi, Bile Ductules, Interlobular Ducts, Hepatic Ducts, Bile Duct, Gallbladder, Cystic Duct (i.e., epithelial lining of the entire hepatobiliary system) |
| Mesoderm | Septum Transversum Mesenchyme | Kupffer cells, Hepatic Stellate Cells, Connective Tissue (Glisson's capsule, portal septa), Hematopoietic cells, Endothelial cells of sinusoids |
Congenital Anomalies of the Hepatobiliary System
Congenital anomalies, though rare, can have significant clinical implications due to the vital functions of the liver and biliary tree.
Accessory Hepatic Ducts:
- These are extra bile ducts that drain parts of the liver.
- They are relatively common anatomical variations and usually drain into the common hepatic duct or common bile duct.
- Their presence is clinically significant because they can be inadvertently ligated or damaged during hepatobiliary surgery (e.g., cholecystectomy), leading to bile leaks, strictures, or jaundice.
Duplication of the Gallbladder:
- A rare anomaly where two separate gallbladders are present, each with its own cystic duct.
- Can be intrahepatic or extrahepatic.
- May be asymptomatic or can present with symptoms similar to single gallbladder disease (e.g., cholecystitis, gallstones). Diagnosis is important to avoid leaving one gallbladder behind during surgery.
Biliary Atresia:
A progressive inflammatory obliterative disease of the extrahepatic or intrahepatic bile ducts, leading to bile outflow obstruction.
Extrahepatic Biliary Atresia (EHBA)
- Incidence: Approximately 1 in 15,000 to 18,000 live births.
- Pathology: Progressive fibrosis and obliteration of the common hepatic or common bile duct.
- Clinical Presentation: Presents in the neonatal period with persistent jaundice (beyond 2 weeks of age), dark urine, and pale stools. If untreated, it leads to cirrhosis, liver failure, and death, usually within the first two years of life.
- Treatment: The Kasai portoenterostomy (hepatoportoenterostomy) is the primary surgical treatment, aiming to create a new pathway for bile drainage. It is most effective when performed before 60 days of age. Even with successful Kasai, many patients will eventually require liver transplantation.
Intrahepatic Biliary Atresia/Hypoplasia
- Pathology: Involves the destruction or absence of intrahepatic bile ducts.
- Incidence: Much rarer than EHBA (e.g., 1 in 100,000 births).
- Etiology: Can be genetic, syndromic (e.g., Alagille syndrome), or acquired (e.g., secondary to severe fetal infections like CMV).
- Clinical Course: Can range from relatively benign to lethal, depending on the extent of duct involvement and associated conditions. May present with cholestasis and jaundice.
Clinical Significance of the Portal and Biliary System
Disruptions in these systems are common causes of significant morbidity and mortality.
Portal Hypertension:
- Definition: Abnormally high blood pressure in the portal venous system.
- Common Cause: Most frequently caused by cirrhosis of the liver, which results in increased resistance to blood flow through the liver due to widespread fibrosis and nodule formation. Other causes include pre-hepatic (e.g., portal vein thrombosis) or post-hepatic (e.g., Budd-Chiari syndrome) obstructions.
- Pathophysiology: Increased pressure in the portal system leads to the development of collateral circulation between the portal and systemic venous systems, bypassing the liver.
- Clinical Manifestations (Portal-Systemic Anastomoses):
- Esophageal varices: Enlarged veins in the lower esophagus, prone to rupture and severe gastrointestinal bleeding (hematemesis).
- Caput medusae: Dilated periumbilical veins radiating from the umbilicus.
- Hemorrhoids: Enlarged veins in the rectum and anus.
- Splenomegaly: Enlargement of the spleen due to congestion (hypersplenism).
- Ascites: Accumulation of fluid in the peritoneal cavity.
- Hepatic encephalopathy: Neuropsychiatric syndrome due to accumulation of toxins (e.g., ammonia) that bypass the liver.
Gallstones (Cholelithiasis):
- Definition: Formation of solid concretions (calculi) within the gallbladder or bile ducts.
- Composition: Most commonly composed of cholesterol or bilirubin (pigment stones).
- Risk Factors: "Five F's" - Fat, Female, Forty, Fertile (multiparity), Fair (Caucasian). Rapid weight loss, certain medications, and genetic factors.
- Clinical Significance:
- Biliary Colic: Episodic, severe abdominal pain (typically in the right upper quadrant) due to temporary obstruction of the cystic duct by a gallstone, often post-prandially.
- Cholecystitis: Inflammation of the gallbladder, usually due to sustained obstruction of the cystic duct by a gallstone, leading to bacterial overgrowth.
- Jaundice: If a gallstone obstructs the common bile duct (choledocholithiasis), it prevents bile flow into the duodenum, leading to cholestasis, accumulation of bilirubin in the blood, and yellowing of the skin and sclera.
- Pancreatitis: If a gallstone obstructs the hepatopancreatic ampulla (Ampulla of Vater), it can cause reflux of bile into the pancreatic duct, leading to acute pancreatitis.