Diabetes Mellitus Type 1
Diabetes Mellitus is defined as a metabolic disorder of multiple etiologies characterized by chronic hyperglycemia with disturbances of carbohydrate, fat, and protein metabolism. This state results from defects of insulin secretion, insulin action, or a combination of both.
Elaboration: Insulin is the primary anabolic hormone of the body. When it is absent or ineffective, the body cannot utilize glucose for energy, leading to a catabolic state where fats and proteins are inappropriately broken down, ultimately resulting in the classic symptoms and potentially life-threatening complications.Historically, diabetes was classified primarily by its treatment modality and age of onset:
- Type 1: Insulin-dependent Diabetes Mellitus (IDDM).
- Type 2: Non-Insulin-dependent Diabetes Mellitus (NIDDM). This was further subdivided into obese and non-obese categories.
- MODY: Maturity-Onset Diabetes of the Young (typically presenting between 18 to 25 years of age).
- IGT: Impaired Glucose Tolerance.
- Gestational Diabetes Mellitus: Diabetes diagnosed during pregnancy.
The modern classification scheme is based on etiology (the underlying cause), rather than on the type of treatment required or the age of the patient at presentation.
- Type I: Characterized by Beta cell destruction leading to absolute insulin deficiency. It is subdivided into:
- Immune-mediated: Autoimmune destruction of the pancreatic beta cells.
- Idiopathic: Beta cell destruction with no known etiology or evidence of autoimmunity.
- Type II: Ranges from predominantly insulin resistant with relative insulin deficiency to a predominant secretory defect accompanied by insulin resistance.
Beyond Type 1 and Type 2, hyperglycemia can result from specific, identifiable secondary causes:
- Genetic defect of beta cell function: Includes MODY syndromes and mitochondrial mutations.
- Infections: E.g., Congenital Rubella and Cytomegalovirus (CMV), which can directly damage the pancreas or trigger an autoimmune response.
- Disease of the Exocrine Pancreas: Any process that diffusely injures the pancreas can cause diabetes, including Pancreatitis, Trauma/Pancreatectomy, Neoplasia, and Cystic Fibrosis.
- Endocrinopathies: Hormonal disorders that produce excess counter-regulatory hormones, such as Acromegaly (excess growth hormone), Cushing's Syndrome (excess cortisol), and Pheochromocytoma (excess catecholamines).
- Drug or Chemical Induced: Medications such as Nicotinic acid, Glucocorticoids, and Thiazides can severely impair insulin action or secretion.
- Genetic Syndromes associated with Diabetes: Increased incidence is seen in Down syndrome, Turner syndrome, Klinefelter syndrome, and Prader-Willi syndrome.
- Gestational Diabetes Mellitus & Neonatal Diabetes Mellitus.
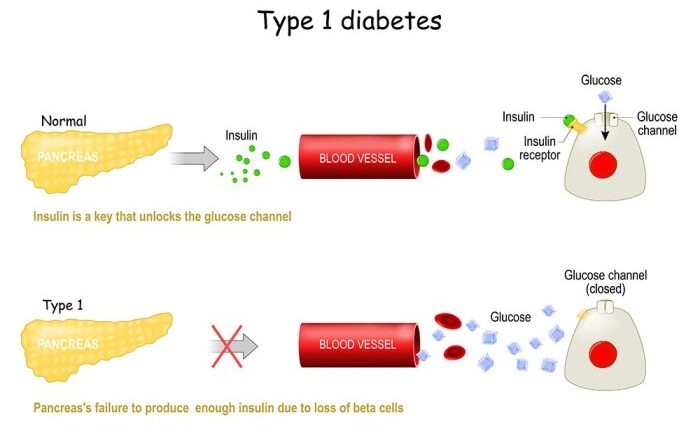
Formerly called insulin-dependent diabetes mellitus (IDDM) or "juvenile diabetes," T1DM is characterized by low or absent levels of endogenously produced insulin due to the targeted destruction of pancreatic beta cells.
- It is the most common endocrine disorder of childhood and adolescence.
- Onset: Occurs predominantly in childhood, featuring a bimodal distribution with two peaks: one at 5-7 years of age (coinciding with increased exposure to infectious agents upon starting school), and another at puberty (coinciding with the physiological stress and counter-regulatory hormone surge of the pubertal growth spurt). However, it may present at any age.
- Prevalence: Varies globally; for instance, in India, an average prevalence of Type I diabetes is estimated at 10 per 100,000 population.
While most cases occur sporadically, there is a clear genetic component to the risk of developing Type 1 DM:
| Relationship to Index Case | Estimated Risk of Developing T1DM |
|---|---|
| If Mother has T1DM | 2% |
| If Father has T1DM | 7% |
| Sibling of index case | 6% |
| Dizygotic (Fraternal) Twins | 6% - 10% |
| Monozygotic (Identical) Twins | 30% - 65% |
The natural history of Type 1 Diabetes involves an interplay between genetic susceptibility, environmental triggers, and immune dysregulation, progressing through distinct stages:
- Genetic Susceptibility: The baseline genetic predisposition.
- Exposure to Environmental "Triggers": Viral infections, dietary factors, or other unknown events that act as a catalyst.
- Initiation of Autoimmunity: The immune system begins to mistakenly target the beta cells in the Islets of Langerhans. Autoantibodies such as IAA (Insulin Autoantibodies), GADA (Glutamic Acid Decarboxylase Autoantibodies), and ICA (Islet Cell Autoantibodies) become positive.
- Preclinical Autoimmunity (Progressive Loss of β-cell function): As beta-cell mass declines over time, the body first loses its first-phase insulin response (the rapid burst of insulin immediately after eating). During this phase, glucose intolerance begins, but fasting blood sugars may remain normal.
- Onset of Clinical Disease (Overt Diabetes): Once approximately 80-90% of the beta-cell mass is destroyed, the remaining cells can no longer maintain normoglycemia, leading to clinical symptoms and the absence of C-peptide (a marker of endogenous insulin production).
- Transient Remission ("Honeymoon Period"): Shortly after initiating exogenous insulin therapy, the few remaining functional beta cells may temporarily recover and secrete insulin, drastically reducing exogenous insulin requirements. This period is transient and eventually followed by complete beta-cell failure.
- Established Disease: Absolute insulin deficiency requiring lifelong exogenous insulin.
- Development of Complications: Arising years later due to chronic microvascular and macrovascular damage.
- DKA (Diabetic Ketoacidosis): The most common initial presentation in pediatrics, presenting as a life-threatening medical emergency.
- Classical Symptom Triad:
- Polyuria: Excessive urination due to osmotic diuresis caused by glycosuria.
- Polydipsia: Excessive thirst due to dehydration from polyuria.
- Weight Loss: Despite normal or increased appetite (polyphagia), the body breaks down fat and muscle for energy due to the inability to utilize glucose.
- Accidental Diagnosis: Detected incidentally during routine urinalysis (glycosuria) or blood tests for other illnesses.
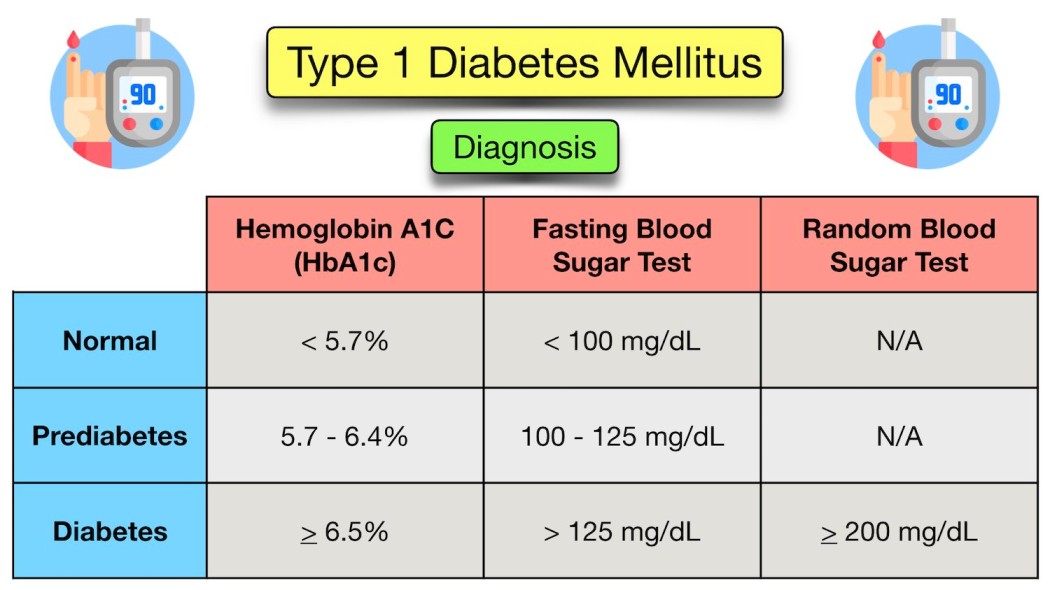
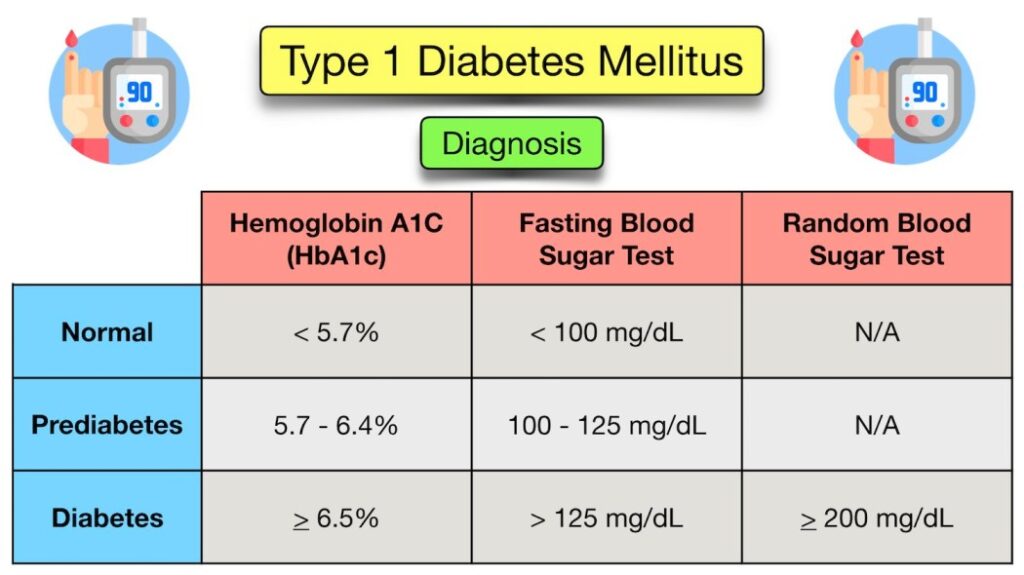
Diagnosis requires clear laboratory evidence of hyperglycemia.
- Symptomatic Children: In children presenting with classic symptoms (polydipsia, polyuria, weight loss), a single Random Plasma Glucose > 11.1 mmol/L (200 mg/dL) is diagnostic.
- Hemoglobin A1c: An HbA1c level of >= 6.5% is universally recognized as diagnostic criteria.
- Asymptomatic or Equivocal Cases: A modified Oral Glucose Tolerance Test (OGTT using 1.75g/kg oral glucose, max 75g) may be needed. This is indicated for asymptomatic children with hyperglycemia (RBS >140) or symptomatic children with borderline hyperglycemia (RBS between 140 to 200).
| Test Parameter | IGT (Impaired Glucose Tolerance) / Pre-Diabetes | Diabetic Threshold |
|---|---|---|
| Fasting Blood Glucose (FBG) | 6.0 - 6.9 mmol/L (100 - 125 mg/dL) | >= 7.0 mmol/L (126 mg/dL) |
| 2 Hours post-OGTT | 7.8 - 11.0 mmol/L (140 - 199 mg/dL) | >= 11.1 mmol/L (200 mg/dL) |

The successful management of T1DM is multifaceted and requires a dedicated team approach involving the physician, diabetes educator, dietitian, and the family.
- Education
- Insulin therapy
- Glycemic control Monitoring
- Diet and meal planning
- Prevention and early detection of complications
Educating the child and caregivers is the cornerstone of management. Key topics include:
- Understanding the pathophysiology of Diabetes Type 1.
- Acceptance of life-long insulin therapy and proper injection techniques.
- Self-monitoring of blood glucose (SMBG) and maintaining accurate logs/records.
- Recognition, prevention, and emergency management of Hypoglycemia & DKA.
- Understanding the meal plan and carbohydrate counting.
- Sick-day management protocols.
- Awareness of possible long-term complications to encourage compliance.
Insulin is a polypeptide made of two beta-chains, famously discovered by Banting & Best in 1921. Historically, animal types (porcine & bovine) were utilized before the introduction of highly purified human-like insulin via DNA-recombinant technology. Recently, more potent insulin analogs have been produced by changing specific amino acid sequences to alter absorption kinetics.
| Insulin Type & Examples | Onset | Peak Time | Duration |
|---|---|---|---|
| Rapid-acting Insulin: Insulin lispro (Humalog), Insulin aspart (Novolog), Apidra |
Begins to work at about 5 - 15 minutes. | Peaks at about 1 - 2 hours. | Continues to work for about 2 - 5 hours. |
| Regular or Short-acting Insulin: Soluble Human Insulin (Actrapid, Humulin R) |
Reaches the bloodstream within 30 minutes after injection. | Peaks anywhere from 2 - 4 hours. | Effective for approximately 3 - 8 hours. |
| Intermediate-acting Insulin: Isophane Insulins: NPH (Insulatard), Lente |
Reaches the blood stream about 2 to 4 hours after injection. | Peaks 4 - 12 hours later. | Effective for about 12 to 18 hours. |
| Long-acting Basal Analogues: Insulin glargine (Lantus), Detemir (Levemir) |
Reaches the bloodstream ~2 to 6 hours after injection. | No pronounced peak ("peakless"). | Usually effective for 18 - 24 hours. Provides a steady basal rate. |
| Pre-mixed Insulins: Mixtard 30, Humulin M3, Novo Mix 30 |
Combines rapid/short acting with intermediate acting. E.g., Mixtard 30 means 30% Soluble (Regular) insulin and 70% Isophane (NPH). Kinetics depend on the mixture ratio. | ||
- Aim: To perfectly mimic the natural, physiological pattern of insulin secretion (a steady basal rate to suppress hepatic glucose output, accompanied by meal-time boluses).
- Concentrations: Insulin is available in 40, 80, & 100 Unit/ml concentrations. To prevent fatal dosage confusion, the WHO now strongly recommends U-100/ml as the only standard formulation. Special high-concentration preparations (U-500/ml) exist for specific profound insulin resistance scenarios.
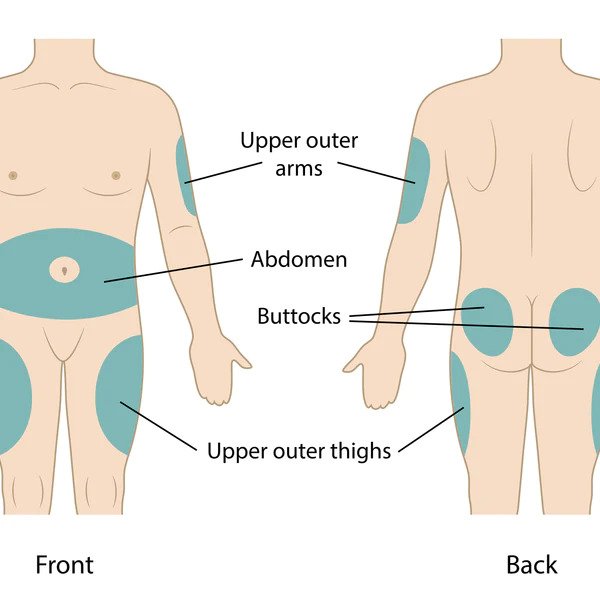
- Administration Sites: Administered subcutaneously using syringes, pens, or pumps. Sites include:
- Anterolateral thighs
- Anterior and lateral abdominal wall
- Posterior aspect of upper arms
- Superolateral aspects of buttocks
Initial Dose Estimation:
- DKA / Overt symptoms: Total Daily Dose (TDD) = 0.8 to 1.0 unit/kg/day.
- Incidentally diagnosed (lower starting doses required):
- Toddlers & pre-school (2-5 yrs): 0.2 - 0.4 unit/kg/day
- Pre-pubertal children (5-9 yrs): 0.5 - 0.8 unit/kg/day
- Adolescents (9-14 yrs): 0.8 - 1.5 unit/kg/day (higher due to pubertal resistance).
Common Regimens:
- Split-Mix Regime (Twice Daily):
- Typically utilizes Mixtard 30:70, OR a manual mix of NPH (2/3) + Regular insulin (1/3).
- Given twice daily: 2/3 of the TDD given 45 mins Before Breakfast (BBF), and 1/3 of the TDD given 45 mins Before Dinner (BD).
- Rule of thumb correction: 1 IU of Mixtard typically takes care of blood sugar ~50mg/dl above target.
- Basal-Bolus Regime (Multiple Daily Injections - MDI):
- Provides superior physiological matching.
- 30-50% of the TDD is given as one dose of long-acting basal insulin (Glargine, Detemir).
- The remainder is divided into 3-4 doses of rapid-acting insulin given right before meals.
- Carbohydrate to Insulin Ratio (CIR): Indicates the amount of carbohydrate in grams covered by one unit of insulin.
- Initial calculation rule: 500 / Total Daily Dose (TDD).
- Accurate estimation: Based strictly on the amount of carbohydrate consumed, units administered, and pre/post-prandial glucose logging.
- Insulin Sensitivity Factor (ISF): Represents the expected reduction in blood glucose by one single unit of rapid-acting insulin.
- Initial calculation rule: 1800 / Total Daily Dose (TDD).
- Correction Formula: For correcting pre-meal high sugars: (Actual BS - Target BS) / ISF = Correction Dose required.
Example Calculation
Patient: 10-year-old child, weighing 33 kg.
Total Insulin Requirement (TDD): 0.8 IU/kg * 33 kg = ~26 IU/day.
CIR (Carb Ratio): 500 / 26 = ~20 grams per unit.
ISF (Correction Factor): 1800 / 26 = ~70 mg/dL per unit.
Basal Regimen: Inj Glargine 40% of TDD = ~11 IU daily.
Scenario: If the child is taking 80g of carbohydrates for lunch, the base meal insulin needed is 80 / 20 (CIR) = 4 IU.
If their pre-lunch blood sugar is elevated at 200 mg/dL (Target is 130), the correction dose is (200 - 130) / 70 (ISF) = 1 IU.
Total Meal Dose: 4 IU (for carbs) + 1 IU (for correction) = 5 IU of Rapid (Lispro) insulin given before lunch.
Continuous Subcutaneous Insulin Infusion (CSII) via battery-powered pumps provides a closer approximation of normal physiological plasma insulin profiles. It accurately delivers a small baseline continuous infusion of rapid-acting insulin (basal rate), coupled with customizable parameters for bolus therapy. The bolus insulin delivery is precisely determined by the amount of carbohydrate intake programmed by the user, cross-referenced with their real-time blood sugar level.
Consistent monitoring ensures therapy is effective and mitigates complications.
- Self Monitoring of Blood Glucose (SMBG): Routine testing should occur fasting, before meals, 2 hours after meals, and during the night.
- Real-time Continuous Glucose Monitoring (CGM): Subcutaneous sensors providing minute-by-minute trending.
- Glycosylated Hemoglobin (HbA1C): Provides an average reflection of glycemic control over the preceding 2-3 months. Must be tested every 3-4 months.
- Measuring Ketones in Urine: More sensitive and accurate for detecting early deterioration. Must be checked when Blood Glucose > 250 mg/dl, or during illness with fever, vomiting, abdominal pain, polyurea, drowsiness, or rapid breathing.
- Urinary Glucose: A very crude indicator of hyperglycemia. It only reflects the glycemic level over the preceding several hours and is only positive if the renal threshold (usually ~180 mg/dL) is exceeded.
| Suggested Target Blood Glucose Range | |
|---|---|
| Time of Checking | Target Plasma Glucose (mg/dL) |
| Fasting or preprandial | 90 - 145 |
| Postprandial | 90 - 180 |
| Bedtime | 120 - 180 |
| Nocturnal | 80 - 162 |
- Adverse Effects: Hypoglycemia (most dangerous acute risk), Lipoatrophy (loss of subcutaneous fat at injection site), Lipohypertrophy (fat accumulation at injection site), Obesity/Weight gain (due to insulin's anabolic nature), Insulin allergy, and development of Insulin antibodies.
- Practical Problems: Non-availability of insulin in poorer nations, poor injection techniques/site rotation, difficulties with insulin storage (cold chain) & transfer, errors in mixing insulin preparations, managing insulin scheduling during school hours, difficulties in adjusting the dose at home, complex sick-day management, and failure to recognize or treat hypoglycemia accurately.
- Regular meal plans utilizing calorie exchange options are highly encouraged.
- Macronutrients: 50-60% of required energy should be obtained from complex carbohydrates. A diet low in salt, low in saturated fats, and high in fiber is heavily encouraged.
- Distribution: Distribute the carbohydrate load evenly during the day, preferably structured as 3 main meals & 2 snacks, with absolute avoidance of simple sugars.
- Split-Mix Regime: Requires strict meal timing. 6 meals daily: 3 major meals (70% of total calories) and 3 mid-meals/snacks (30% of total calories).
- MDI Regime: Offers more flexibility. Mid-meals are not essential as the basal covers fasting periods. If taken, mid-meals should have less than 10-15 gm of carbohydrate.
- Glycemic Index (GI): A ranking of carbohydrates on a scale from 0 to 100 according to how rapidly they raise blood sugar levels.
- High GI: Rapidly digested, causing marked blood sugar spikes (e.g., corn flakes, potato, watermelon, biscuits, chocolates). Should be avoided.
- Low GI: Slow digestion and absorption, producing gradual rises in blood sugar and insulin levels. Proven benefits for diabetics (e.g., most fruits, non-starchy vegetables, pasta, pulses, milk, curd).
Exercise is a fundamental pillar of therapy. It profoundly decreases insulin requirements by increasing both the sensitivity of muscle cells to insulin and the direct, non-insulin-mediated utilization of glucose by skeletal muscle. However, because the body cannot shut off injected insulin, exercise can easily precipitate severe hypoglycemia in an unprepared diabetic patient if carbohydrates are not supplemented prior to activity.
Managing diabetes during acute illnesses (colds, flu, gastroenteritis) is critically challenging. Insulin requirements may unexpectedly increase or decrease.
- Stress Physiology: Fever, dehydration, and the physiological stress of illness cause hyperglycemia due to increased production of counter-regulatory hormones (cortisol, glucagon, epinephrine). Conversely, vomiting and loss of appetite can lead directly to hypoglycemia.
- Ketosis Risk: The risk of ketosis is exponentially increased due to the combination of starvation (not eating) and dehydration.
- Action Plan:
- Take plenty of fluids to prevent dehydration.
- Blood glucose and urine ketones must be monitored frequently (every 2-4 hours).
- The presence of "moderate" or "large" ketones in the urine alongside hyperglycemia is a dangerous red flag, indicating severe insulin deficiency and high risk for impending DKA.
- The child should be given additional correction doses of rapid-acting or regular insulin (e.g., 0.1 u/kg if RBS > 250), alongside oral fluids. Ketones must be rechecked in the next urine output.
- Red Flag: If there is persistent vomiting with hyperglycemia and large ketones, or uncontrollable persistent hypoglycemia, the child must be taken to the emergency department immediately.
- Delayed diagnosis resulting in presentation at the DKA stage.
- Misinterpreting the "Honeymoon period" as a cure.
- Failure to properly diagnose and treat DKA and hypoglycemia.
- Dawn Phenomenon vs. Somogyi Phenomenon: These conditions both result in elevated morning glucose but require opposite treatments.
- Dawn Phenomenon: Blood glucose levels steadily increase in the early morning hours before breakfast. This is a normal physiologic process caused by the overnight surge of growth hormone secretion and increased clearance of insulin. While non-diabetics simply secrete more insulin to compensate, a T1DM child cannot, resulting in morning hyperglycemia. Treatment: Increase the evening basal insulin.
- Somogyi Phenomenon: A theoretical rebound hyperglycemia. It occurs when a patient experiences undetected late-night or early morning hypoglycemia (e.g., from too much evening insulin). The body panics and mounts an exaggerated counter-regulatory hormone response (glucagon, epinephrine), heavily dumping glucose into the blood, leading to ambiguously elevated morning glucose levels. Treatment: Decrease the evening basal insulin or provide a bedtime snack. Continuous glucose monitoring or 3:00 AM fingersticks clarify which phenomenon is occurring.
- Hypoglycemia: Defined as blood glucose < 70 mg/dL. The risk increases as the duration of diabetes increases.
- Mild to moderate: Immediate oral intake of 0.3 g/kg of fast-acting glucose dissolved in a small amount of water (raises blood glucose by 45-65 mg/dL). Retest after 10-15 minutes and readminister if the response is inadequate. Note: Chocolate, milk, and fat-containing sweets are poor choices for emergency rescue as fat significantly delays the gastric emptying and absorption of glucose. Avoid treating with the child's favorite candies to prevent behavioral reinforcement.
- Severe (Altered mental status, unconsciousness, or seizures): Glucagon is administered subcutaneously or intramuscularly. DOSE: 0.5 mg for < 12 years; 1.0 mg for > 12 years. If glucagon injections are unavailable, glucose gel or honey can be carefully rubbed into the buccal pouch.
- Diabetic Ketoacidosis (DKA): A life-threatening complication occurring more commonly in children with Type 1 DM than Type 2 DM.
- Definition: Hyperglycemia (serum glucose > 200-300 mg/dL) combined with severe metabolic acidosis (blood pH < 7.3, serum bicarbonate < 15 mEq/L) and prominent ketonemia/ketonuria.
- Signs & Symptoms: Nausea, vomiting, severe abdominal pain, fruity (acetone) odor on breath, tachycardia, low volume pulses, hypotension, impaired skin turgor, delayed capillary refill, profound dehydration, and rapid, deep sighing respirations known as Kussmaul respirations (a respiratory compensation for the metabolic acidosis).
Classification of Diabetic Ketoacidosis Parameter Normal MILD DKA MODERATE DKA SEVERE DKA Venous CO2 (mEq/L) 20 - 28 16 - 20 10 - 15 < 10 Venous pH 7.35 - 7.45 7.25 - 7.35 7.15 - 7.25 < 7.15 Clinical State No change Oriented, alert but fatigued Kussmaul respirations; oriented but sleepy; arousable Kussmaul or depressed respirations; sleepy to depressed sensorium leading to coma DKA Treatment Protocol:- 1st Hour: Quick volume expansion. 10-20 ml/kg IV bolus of 0.9% NaCl (Normal Saline) or Lactated Ringer's. May be repeated. Keep patient NPO. Monitor I/O and neurological status closely. Have Mannitol at the bedside (1 g/kg IV push) ready for cerebral edema. Initiate continuous Insulin drip at 0.05 to 0.10 units/kg/hr.
- 2nd Hour until DKA resolution: Change fluids to 0.45% NaCl. Continue Insulin drip. Crucially, when blood sugar drops below 250 mg/dL, add 5% glucose to the IV fluid to prevent hypoglycemia while insulin continues to clear ketones. Add 20 mEq/L Potassium Phosphate (If K < 3 mEq/L, give 0.5 to 1 mEq/kg as oral K or increase IV K to 80 mEq/L). Total IV rate should equal roughly 85 ml/kg + maintenance - bolus divided by 23hr.
- Transition to Subcutaneous: Occurs after correction of dehydration and acidosis (No emesis, CO2 > 16, normal electrolytes). As oral feeds advance, IV fluids are reduced. The ideal time to transition is just before a meal. Rapid-acting insulin is given 15-30 mins prior, and regular insulin 1-2 hours prior to stopping the IV infusion to prevent rebound hyperglycemia.
- Cerebral Edema: The most dreaded, fatal complication of DKA treatment, often caused by rapid fluid shifts.
- Management: Head end elevation. Give Mannitol 0.5-1 gm/kg immediately and repeat if no response in 30 mins to 2 hrs. 3% Hypertonic saline (5 ml/kg over 30 mins) can alternatively be given. Restrict IV fluids to 2/3 maintenance. Replace the remaining fluid deficit slowly over 72 hours rather than 24 hours. Intubation and hyperventilation may be required.
- Non-Ketotic Hyperosmolar Coma (NKHS): Uncommon in children, but exceptionally dangerous.
- Features: Severe, massive hyperglycemia (blood glucose > 800 mg/dL), absence of (or only slight) ketosis, nonketotic acidosis, profound, severe dehydration, depressed sensorium or frank coma, and variable neurological signs (grand mal seizures, hyperthermia, hemiparesis, positive Babinski signs). Respirations are shallow. Serum osmolarity > 350 mOsm/kg.
- Treatment: Requires rapid repletion of the vascular volume deficit, but very slow correction of the hyperosmolar state to avoid cerebral edema. 0.45% NaCl is administered (replacing 50% deficit in 1st 12hr, remainder over next 24hr). When glucose approaches 300 mg/dL, change to 5% dextrose in 0.2 NS. Insulin drip is lower (0.05 units/kg/hr) and delayed until the 2nd hour of fluid therapy. Aggressive potassium replacement (20 mEq/L) is required.

Chronic hyperglycemia damages blood vessels over decades, leading to microvascular and macrovascular disease.
- Retinopathy: Damage to the retina leading to blindness. Screening: Fundal photography. Begin 5 years post-diagnosis in prepubertal children, and 2 years post-diagnosis in pubertal children. Frequency: 1-2 yearly.
- Nephropathy: Kidney damage progressing to renal failure. Screening: Spot urine sample for albumin:creatinine ratio (detecting microalbuminuria). Timing: Same as retinopathy. Frequency: Annually.
- Neuropathy: Nerve damage causing numbness, tingling, and pain. Screening: Physical examination (monofilament test). Screening timelines are less clear in children, but typically assessed 5 years after T1DM diagnosis.
- Macrovascular Disease: Ischemic heart disease and stroke. Screening: Fasting lipid profile. Starts after age 2. Frequency: Prepubertal every 5 years; Pubertal within 6-12 months of diagnosis, then every 2 years.
- Autoimmune Co-morbidities: T1DM patients frequently develop other autoimmune diseases.
- Thyroid Disease: Screen via TSH at diagnosis, then every 2-3 years.
- Celiac Disease: Screen via tissue transglutaminase (tTG) and endomysial antibodies at diagnosis, then every 2-3 years.
- Pancreas & Islet Cell Transplantation: Whole pancreas transplants are typically reserved for diabetics suffering from end-stage renal disease who are already receiving kidney transplants (and thus already on immunosuppressants). Islet cell transplants (the ultimate theoretical treatment) are under trials globally with encouraging results, but graft rejection and the recurrence of the underlying autoimmunity are serious, persistent limitations.
- Immune Modulation: Utilizing immunosuppressive therapy for newly diagnosed patients or high-risk children to prolong the "honeymoon phase" and preserve remaining beta cells. Drugs investigated include Nicotinamide and Mycophenolate.
- Gene Therapy: Cutting-edge research attempting to block the immunologic attack against islet cells using DNA-plasmids encoding self-antigens, genes encoding cytokine inhibitors, or modifying gene-expressed islet-cell antigens like GAD.
Prediction: Evaluating high-risk individuals utilizing sensitive and specific immunologic markers such as GAD Antibodies, GLIMA antibodies, and IA-2 antibodies. Having a single antibody presents a 2-6% risk of developing T1DM, two antibodies increase risk to 21-40%, and carrying more than two antibodies creates a 59-80% definitive risk.
Prevention Tiers:
- Primary Prevention: Intervening before any disease process starts. Includes identification of the diabetes gene, theoretical tampering/re-education of the immune system, and elimination of triggering environmental factors.
- Secondary Prevention: Intervening after antibodies appear but before overt clinical diabetes. Utilizes immunosuppressive therapies (e.g., cyclosporin) and GLP-1 agonists to delay onset.
- Tertiary Prevention: Focuses on established disease. It demands tight metabolic control and excellent monitoring to prevent the devastating late-onset complications.
Diabetes Mellitus Type 1 Read More »