Hyperparathyroidism and Hypoparathyroidism
The parathyroid glands are critical regulators of calcium, phosphate, and bone metabolism. Pathologies affecting these glands typically manifest as disorders of calcium homeostasis, presenting either as hypercalcemia (hyperparathyroidism) or hypocalcemia (hypoparathyroidism).
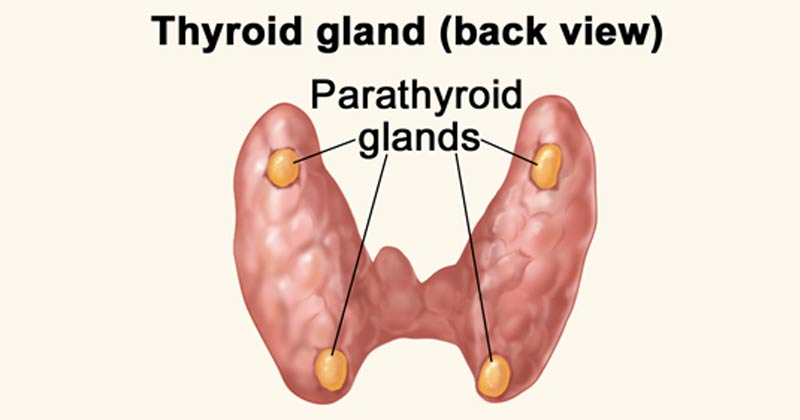
- Definition: Small endocrine glands located in the neck, usually situated behind the thyroid gland, responsible for producing parathyroid hormone (PTH).
- Embryology: They develop early in gestation at approximately 6 weeks and migrate caudally by 8 weeks. The inferior glands derive from the 3rd pharyngeal pouch, while the superior glands derive from the 4th pharyngeal pouch.
- Number: There are typically 4 parathyroid glands (two superior and two inferior).
- Ectopic Locations: Because of their embryological migration, glands can sometimes be found in unusual locations.
- Paraesophageal (28%)
- Mediastinum (26%)
- Intrathymic (24%)
- Intrathyroidal (11%) - located entirely within the thyroid gland capsule.
- Carotid sheath (9%)
- High cervical (2%)
- 1849: Sir Richard Owen provided the 1st accurate description of normal parathyroid glands after examining an Indian Rhinoceros.
- 1879: Anton Wölfler described tetany in a patient after a total thyroidectomy, hinting at the glands' function.
- 1880: Ivar Sandström, a Swedish medical student, grossly and microscopically described the parathyroid glands in humans.
- 1909: Calcium measurement became possible, and its firm association with the parathyroids was established.
- 1925: The 1st successful parathyroidectomy was performed on a 38-year-old man suffering from severe bone pain secondary to osteitis fibrosa cystica.
PTH is synthesized in the chief cells of the parathyroid gland as a larger precursor molecule. It undergoes sequential cleavage:
- Preproparathyroid hormone (the initial synthesized form).
- Cleaved into proparathyroid hormone.
- Cleaved finally into the active 84-amino-acid PTH.
Secreted PTH has an extremely short half-life of 2 to 4 minutes. In the liver, PTH is metabolized into an active N-terminal component and a relatively inactive C-terminal fraction.
Clinical Note: The short half-life of PTH is highly advantageous during parathyroidectomy surgery. Surgeons can measure intraoperative PTH levels; a drop of >50% within 10 minutes of removing a suspicious gland confirms the successful removal of the hypersecreting adenoma.The overarching goal of PTH is to increase serum calcium levels and decrease serum phosphate levels. It achieves this via three main target organs:
- Bone: Activates and increases the number of osteoclasts, which mobilizes (resorbs) calcium and phosphate from bone tissue into the blood.
- Kidneys:
- Increases renal tubular reabsorption of calcium (preventing its loss in urine).
- Increases urinary phosphate excretion (phosphaturic effect), which prevents calcium-phosphate precipitation and effectively raises free ionized calcium.
- Increases the conversion of Vitamin D to its most active form, 1,25-dihydroxyvitamin D3 (Calcitriol).
- Gastrointestinal Tract: Indirectly increases GI calcium absorption via the actions of the newly activated Vitamin D.
The interaction between PTH and bone cells is complex and indirect:
- PTH stimulates the osteocytic pump, increasing the permeability of the osteocytic membrane, allowing calcium to diffuse rapidly from bone fluid into the blood.
- Receptor Specificity: Osteoblasts and osteocytes have PTH receptors. Osteoclasts do not have PTH receptors.
- Signaling Mechanism: PTH stimulates osteoblasts/osteocytes, which then activate osteoclasts via a complex "signaling" system (physiologically known as the RANK/RANKL pathway). Therefore, PTH indirectly stimulates the formation of new osteoclasts.
- Net Effect: While both osteoblastic (building) and osteoclastic (destroying) cell lines are activated by PTH, the clastic activity > blastic activity, resulting in net bone resorption.
Calcitonin is a 32-amino-acid-long peptide produced by the parafollicular cells (C cells) of the thyroid interstitium. It acts as the physiological antagonist to PTH.
- Action: Temporarily lowers serum calcium levels.
- Mechanism: Decreases osteoclastic bone resorption activity and stimulates a distal tubular-mediated calciuresis (calcium excretion in the kidney).
- Stimulus: Secretion is directly stimulated by high blood calcium levels.
Before assuming hyperparathyroidism, it is critical to understand that approximately 80% of all hypercalcemia cases are caused by either Malignancy or Primary Hyperparathyroidism. The mnemonic VITAMINS TRAP helps recall the various causes:
| Letter | Etiology | Letter | Etiology |
|---|---|---|---|
| V | Vitamins (A and D intoxication) | T | Thiazide diuretics, other drugs (Lithium) |
| I | Immobilization (prolonged bed rest) | R | Rhabdomyolysis |
| T | Thyrotoxicosis (Hyperthyroidism) | A | AIDS |
| A | Addison's disease (Adrenal insufficiency) | P | Paget's disease, Parenteral nutrition, Pheochromocytoma, Parathyroid disease |
| M | Milk-alkali syndrome | ||
| I | Inflammatory disorders | ||
| N | Neoplastic related disease (Malignancy) | ||
| S | Sarcoidosis (Granulomatous diseases) |
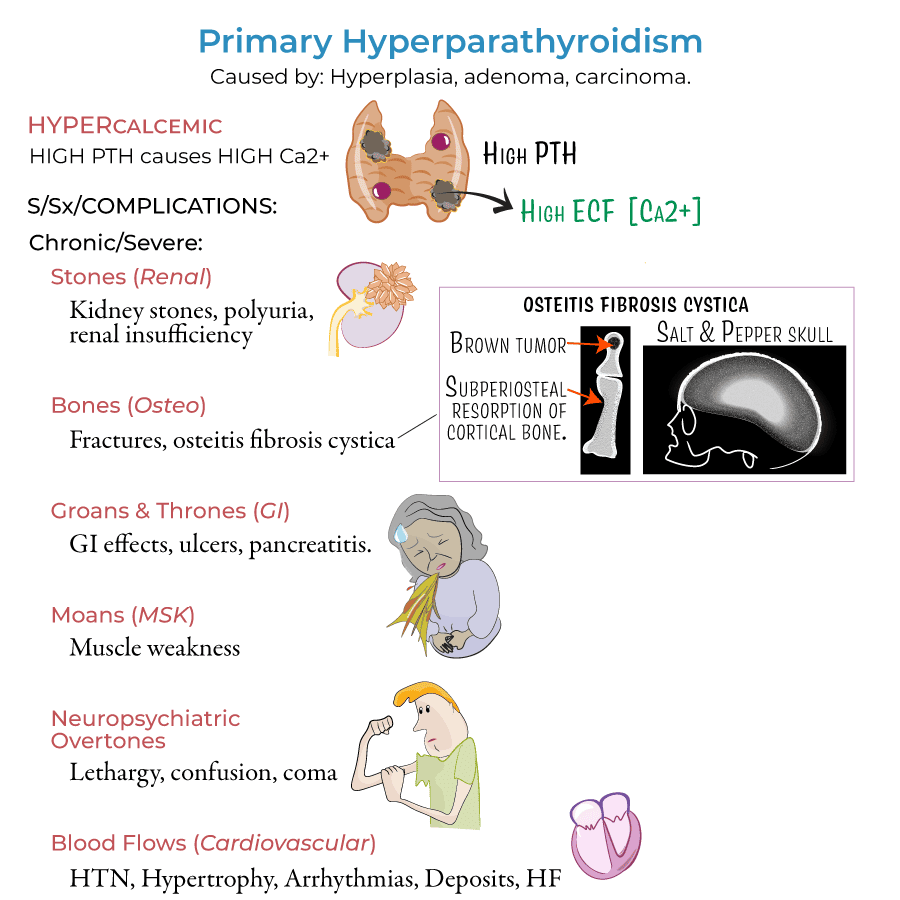
Hyperparathyroidism is characterized by the excessive secretion of PTH, leading to disruptions in calcium and phosphate homeostasis.
- Primary Hyperparathyroidism:
- Definition: The unregulated, autonomous overproduction of PTH resulting in abnormal calcium homeostasis (hypercalcemia).
- Epidemiology: Incidence of 27 cases per 100,000 annually. Prevalence in the general population is 0.1%-0.3%, but it rises to >1% in women over 60 years old. There are approximately 50,000 new cases yearly.
- Etiology:
- Adenoma of a single parathyroid gland (most common, ~80-85%).
- Multiple adenomas.
- Parathyroid Hyperplasia (all 4 glands enlarged).
- Parathyroid Carcinoma (rare, <1%).
- Secondary Hyperparathyroidism:
- Definition: The overproduction of PTH secondary to a chronic abnormal stimulus for its production. It is a compensatory mechanism to maintain calcium levels in the face of a calcium-depleting condition.
- Etiology:
- Chronic Kidney Disease (CKD): Failing kidneys cannot excrete phosphate or activate Vitamin D. Hyperphosphatemia appears to be particularly important in the development of parathyroid hyperplasia.
- Vitamin D Deficiency: Decreased calcium absorption from the intestine leads to chronic hypocalcemia, stimulating the parathyroid glands.
- Pathophysiology in CKD: Decreased active Vitamin D and increased phosphate lower serum ionized calcium -> Parathyroid glands undergo hyperplasia to pump out more PTH -> Result is high PTH, normal/low Calcium, and severe bone disease (renal osteodystrophy).
- Tertiary Hyperparathyroidism:
- Definition: Characterized by the development of autonomous hypersecretion of PTH causing hypercalcemia, occurring after long-standing secondary hyperparathyroidism.
- Etiology: The exact etiology is unknown, but a change may occur in the set point of the calcium-sensing mechanism to hypercalcemic levels. The hyperplastic glands simply stop responding to negative feedback, even if the underlying cause (like kidney failure) is corrected (e.g., post-renal transplant).
The classical presentation of hyperparathyroidism is memorized by the rhyme: "Stones, Bones, Groans, and Moans."
- Muscular weakness and severe fatigue.
- Poor appetite, anorexia, vomiting, constipation, and loss of weight.
- Slow and/or shaken "duck" gait.
- Polyuria, polydipsia (nephrogenic diabetes insipidus due to calcium interfering with ADH).
- Hyposthenuria (inability to concentrate urine) and alkaline reaction of urine.
- Nephrolithiasis / Renal calcinosis: Calcium phosphate or calcium oxalate kidney stones (coral stones).
- Renal infections and potential progression to renal failure.
Continuous excessive PTH strips the skeleton of calcium, leading to profound radiological and clinical findings:
- Severe osteoporosis and osteopenia; bone and joint pains.
- Pathological bone fractures.
- Osteitis Fibrosa Cystica: Advanced bone disease with cystic "brown tumors" replacing normal marrow.
- Subperiosteal Resorption: Classical X-ray finding, especially visible on the radial aspect of the middle phalanges of the fingers.
- Pepper-pot skull: Also called "salt and pepper appearance" on skull X-rays due to punctate decalcification.
- Rugger jersey spine: Sclerotic bands on the superior and inferior endplates of vertebrae.
- Dental Changes: Loss of teeth, Epulis (Giant cell tumor/granuloma of the jaw). Loss of lamina dura is a pathognomonic oral change seen on dental panoramas (radiopaque teeth standing out in contrast to radiolucent jaws).
- Peptic ulcer disease and bleeding (calcium stimulates gastrin secretion).
- Calculous pancreatitis.
- Headaches.
- Mental status changes, confusion, depression, lethargy.
- High blood pressure (Hypertension).
- Heart palpitations and arrhythmias.
- Left ventricular hypertrophy.
- Accelerated vascular calcification.
- Band Keratopathy: Deposition of calcium at the corneal/scleral junction. Common in long-standing hypercalcemia. Calcium deposition begins near the limbus at the 3 & 9 o'clock position (where there is less friction from the lids). The tear film is most alkaline in the most exposed area, precipitating calcium as a band running across the cornea.
| Calcium Status | Values |
|---|---|
| Normal Total Serum Calcium | 2.0 to 2.5 mmol/L |
| Normal Ionized Calcium | 1.0 to 1.4 mmol/L |
| Hypercalcemia | Total > 2.5 mmol/L OR Ionized > 1.4 mmol/L |
| Severe Hypercalcemia | Total > 3.5 mmol/L |
A medical emergency present when severe neurological symptoms (coma, stupor) or cardiac arrhythmias are present in a patient with a serum calcium > 3.5 mmol/L, or automatically when the serum calcium is > 4.0 mmol/L. ECG findings often include a shortened Q-T interval.
- Clinical Presentation: General weakness, fatigue, bone pain, skeleton deformation, fractures.
- Laboratory Data: Hypercalcemia, hypophosphatemia, hypercalcinuria. Increased levels of Alkaline Phosphatase (reflecting high bone turnover). High intact PTH levels.
- Radiology (Bone Densitometry/DXA): Identifies osteoporosis/osteopenia.
- T-score measures Standard Deviations (SD) from the young adult mean.
- Normal: T above -1.
- Osteopenia: T between -1 and -2.5.
- Osteoporosis: T below -2.5.
- Severe Osteoporosis: T below -2.5 AND fragility fractures.
- Localization Studies (Imaging for Adenoma): Used primarily for surgical planning.
- Ultrasound of the neck.
- Colour Doppler (demonstrates typical vascularity of the adenoma).
- CT scan / MRI: Especially useful for ectopic glands (e.g., identifying an adenoma in the upper mediastinum behind the oesophagus).
In acute severe forms (Hypercalcemic Crises), the mainstay of therapy is immediate reduction of serum calcium:
| Treatment | Onset / Duration | Mechanism & Advantages |
|---|---|---|
| Hydration with Saline | Onset: Hours. Duration: During infusion. | Adequate hydration dilutes serum calcium. Rehydration is invariably needed as patients are usually profoundly volume depleted. |
| Forced Diuresis | Onset: Hours. Duration: During treatment. | Saline plus Loop Diuretic (e.g., Furosemide) increases the rapid urinary excretion of calcium along with sodium, preventing its reabsorption by the renal tubules. (Note: Never use Thiazides as they retain calcium). |
| Bisphosphonates | Onset: 1-2 days. Duration: Days to weeks. | Bind to calcium in bone and completely inhibit osteoclast activity. High potency.
|
| Calcitonin | Onset: Hours. Duration: 1-2 days. | Rapid onset of action. Very useful as a fast adjunct in severe hypercalcemia while waiting for bisphosphonates to take effect. |
| Phosphate | Onset: Hours (IV) to 24h (Oral). | Oral is used for chronic management if hypophosphatemia is present. IV is highly potent but rarely used due to the massive risk of metastatic calcification (cardiac/renal decompensation). |
| Glucocorticoids | Onset: Days. | Oral therapy, particularly useful as an antitumor agent for hypercalcemia of malignancy (e.g., multiple myeloma, lymphomas). |
| Dialysis | Onset: Hours. | Useful in renal failure; can immediately reverse life-threatening hypercalcemia. |
The basic and definitive method of treatment for Primary Hyperparathyroidism is surgical.
- Significant symptoms of hypercalcemia.
- Nephrolithiasis (kidney stones).
- Decreased bone mass (T-score > 2 standard deviations below mean for age, i.e., osteoporosis).
- Serum calcium > 2.5 mmol/L (or >1.0 mg/dL above the upper limit of normal).
- Age < 50 years.
- Infeasibility of long-term medical follow-up.
- Surgeon must explore and find all four glands. Intraoperative frozen sections and rapid PTH measurements are highly useful.
- If a single gland is enlarged (Adenoma): Removal is usually curative.
- If multiple glands are enlarged: They are removed; normal-appearing glands are just biopsied.
- If all 4 are enlarged (Generalized Parathyroid Hyperplasia): A subtotal parathyroidectomy is performed (removing 3 & 1/2 glands). The remaining half-gland is often re-implanted into a reachable muscle bed (like the forearm muscle) to maintain basic PTH levels and allow easy removal if hyperparathyroidism recurs.
Following successful surgery, the bones rapidly pull calcium from the blood to rebuild (a phenomenon known as "Hungry Bone Syndrome"). For the fastest restoration of bone structure, the following are recommended:
- Diet fortified with calcium.
- Calcium medications and Vitamin D3 supplementation.
- Anabolic steroids (to promote tissue building).
- Calcitonin.
- Physiotherapy exercises and massage.
Hypoparathyroidism is the state of decreased secretion or activity of parathyroid hormone (PTH).
Synonyms/Associated terms: Tetany, Hypocalcemia, DiGeorge Syndrome, Osteomalacia, Pseudohypoparathyroidism.
Hypoparathyroidism broadly falls into three pathophysiological categories:
- Deficient PTH secretion: The glands are absent, damaged, or suppressed.
- Inability to make an active form of PTH: Genetic defects in synthesis.
- Inability of the kidneys & bones to respond to PTH: Known as Pseudohypoparathyroidism.
- Characterized by resistance to PTH.
- Patients have normal or high PTH levels but exhibit tissue insensitivity to the hormone.
- Clinically associated with mental retardation, skeletal deformities (e.g., short stature, short 4th and 5th metacarpals), collectively termed Albright Hereditary Osteodystrophy.
- These rare individuals have plenty of PTH, but their organs simply do not behave appropriately to it due to receptor or G-protein defects.
- Post-Surgical: Operations designed to remove parathyroid glands for hyperparathyroidism. Accidental removal or devascularization during a total thyroidectomy or surgery for laryngeal/neck malignancy.
- Extensive Irradiation: Radiation therapy to the face, neck, or mediastinum.
- "Hungry Bone Syndrome": Develops acutely after a parathyroidectomy for severe hyperparathyroidism, as described above.
- May exist alone (sporadic/familial) or as part of a syndrome. Average age of hypocalcemia onset is 7 years.
- Type 1 Autoimmune Polyglandular Syndrome (APS-1): Also referred to as HAM syndrome (Hypoparathyroidism, Addison's, Mucocutaneous candidiasis). This involves the direct autoimmune destruction of the parathyroid glands.
- Iron & Copper Overload: Hemochromatosis and Thalassemia (iron overload) or Wilson disease (copper overload) result in metal deposition in the glands, causing destruction and primary hypoparathyroidism.
- Aluminum Deposition: Occurs within parathyroid glands of end-stage renal disease patients on hemodialysis.
- Magnesium Imbalance: Hypermagnesemia decreases PTH release. Hypomagnesemia causes a reversible, functional primary hypoparathyroidism (magnesium is required for PTH exocytosis and peripheral receptor action).
- Granulomatous diseases (Sarcoidosis).
- Amyloidosis.
- Syphilis.
- Progressive systemic sclerosis.
- Occurs in the unborn baby of a mother with hypercalcemia. The maternal hypercalcemia crosses the placenta and causes chronic suppression of the fetal parathyroid gland function. At birth, the maternal calcium supply is severed, and the newborn is at immediate risk of profound hypocalcemia.
- Branchial Dysgenesis (DiGeorge Syndrome): A failure of the 3rd and 4th pharyngeal pouches to develop, leading to absent thymus and parathyroids, along with cardiac defects.
- Isolated primary hypoparathyroidism (monogenic, autosomal dominant or recessive conditions).
- Diabetic embryopathy.
Low serum calcium lowers the threshold for nerve action potentials, leading to severe neuromuscular hyperexcitability.
- Muscle spasm or cramping (Tetany):
- "Obstetrician Hand" (Carpal Spasm): The hand is fixed with wrist flexed, metacarpophalangeal joints flexed, and interphalangeal joints extended.
- "Tip Foot" (Pedal Spasm): A sharp plantar bending of the foot with bent toes.
- "Sardonic Smile" / "Fish Mouth": Spasm of the facial muscles.
- Trismus: Severe spasm of the chewing (masseter) muscles.
- Convulsions and seizures.
- Paresthesias: Abnormal sensations such as numbness, tingling, or burning, especially noticeable around the mouth (perioral) and fingertips.
- Anxiety.
- Hoarseness & Stridor: Highly dangerous, caused by laryngospasm (spasm of the vocal cords) obstructing the airway.
- Wheezing & Dyspnea: Due to bronchospasm.
- Hypotension and resistance to digitalis drugs.
- Refractory heart failure with cardiomegaly can occur in chronic cases.
- Cataracts (calcium deposition in the lens).
- Hair loss, dry skin, and malformed, brittle nails.
- Poor tooth development in children (hypoplasia of enamel).
- Candidiasis (yeast infection), especially noted in autoimmune polyglandular syndrome.
| Sign | Description |
|---|---|
| Chvostek's (Weiss) Sign | An abnormal spasm of the facial muscles elicited by lightly tapping the patient's facial nerve just anterior to the earlobe (near the lower jaw). |
| Trousseau's Sign | An indication of latent tetany in which a carpal spasm occurs when the upper arm is compressed by a blood pressure cuff inflated above systolic pressure for 3 minutes. (More sensitive and specific than Chvostek's). |
| Schlesinger's Symptom | If the patient's lower limb is held at the knee joint and flexed strongly at the hip joint, there will soon be an extensor spasm at the knee joint, with extreme supination of the foot. |
| Hoffmann's Sign | Increased excitability to electrical stimulation in sensory nerves; typically, the ulnar nerve is tested. |
- Calcium & Phosphorus: Hypocalcemia is characterized by abnormally low levels of calcium and high levels of phosphorous (hyperphosphatemia) in the blood.
- PTH Levels:
- Primary Hypoparathyroidism: Serum PTH is Low (↓), Calcium is Low (↓).
- Pseudohypoparathyroidism: Serum PTH is High (↑), Calcium is Low (↓).
- Secondary Hypoparathyroidism: (e.g. severe Vit D deficiency causing exhaustion of glands): Serum PTH Low/High, Calcium Low.
- Vitamin D: Measurement of 25-hydroxy vitamin D is vital to exclude nutritional vitamin D deficiency as the root cause of the hypocalcemia.
- Magnesium: Serum magnesium must be checked. Hypomagnesemia causes PTH deficiency.
- ECG Findings: Typically shows a prolonged QT interval and various arrhythmias due to delayed ventricular repolarization.
- Monitoring: A blood test every three months is recommended for patients whose serum calcium and symptoms are stable, with more frequent testing for those who are unstable.
- Rich in Calcium: Dairy produce, almonds, legumes, dark leafy greens, blackstrap molasses, oats, sardines, prunes, apricots, and sea vegetables.
- Low in Phosphorus: Must avoid phosphorus-rich items. This specifically means avoiding meat-heavy diets and carbonated soft drinks, which contain high levels of phosphorus in the form of phosphoric acid.
- Oral Calcium Tablets (Calcium Salts):
- Calcium Carbonate (Cal-Plus, Caltrate, Os-Cal 500): 1-2 g/day of elemental calcium PO (which equates to 2.5-5 g/day of actual calcium carbonate compound). It contains 400mg elemental Ca per 1g of salt.
- Calcium Citrate (Citracal): 1-2 g/day elemental calcium PO. (Better absorbed in patients with low stomach acid). It contains 211mg elemental Ca per 1g of salt.
- Vitamin D Preparations: Massive doses are required because PTH is not available to convert inactive Vitamin D to active Calcitriol in the kidney.
- Ergocalciferol (D2): 50,000 - 100,000 U/day PO/IM. (100 times the physiological requirement). Treatment begins with a loading dose of 250,000 - 400,000 units.
- Dihydrotachysterol (DHT): 125 - 250 mcg/d PO.
- Calcifediol: 50 - 220 mcg/d PO.
- Calcitriol (Active D3, Rocaltrol): 0.5 - 1 mcg/day PO. Preferred as it acts rapidly and bypasses the kidney activation step.
- Synthetic form of PTH: Teriparatide.
An acute tetanic attack (especially with laryngospasm) is a life-threatening medical emergency requiring immediate intravenous calcium.
| Phase | Action | Rationale / Details |
|---|---|---|
| Acute Resuscitation | Administer IV Calcium Gluconate. | 10–60 ml of 10% Calcium Gluconate diluted in 50 mL of 5% dextrose (D5W) or 0.9% Normal Saline, given slowly IV over 5 minutes. Note: 1g of Calcium Gluconate provides 90 mg of elemental calcium. |
| Maintenance Infusion | Set up a continuous IV infusion. | Continuing hypocalcemia often requires a constant infusion: typically 10 ampules of calcium gluconate (or 900 mg of elemental calcium) mixed in 1 Liter of 5% dextrose or 0.9% sodium chloride, administered continuously over 24 hours. |
Hyperparathyroidism and Hypoparathyroidism Read More »